The IEC 62304 standard includes requirements for managing the life cycle of medical device software, from design and development to manufacturing, distribution, and post-market surveillance.
This illustrated guide was written by Wasif Ali Syed,
This guide will:
- Provide a useful overview of IEC 62304,
- Clarify terminology used in the standard, and
- Address common misconceptions
What is medical device software?
As an example, let’s use a summarised version of the definition of a medical device from the MDR as a starting point:
“A medical device is any instrument, apparatus, appliance, software, material, or other article intended for use in the diagnosis, prevention, monitoring, treatment, or alleviation of disease or injury. It must achieve its intended purpose primarily through physical or mechanical means, not by pharmacological, immunological, or metabolic means.”
Simply put, ‘medical device software’ refers to any software associated with a medical device. Regardless of the specific technology or terminology, SiMD, SaMD, MDSW, firmware, or embedded software, the intended purpose makes it a medical device software.
The platform doesn’t alter its classification. Whether the software runs on mobile, cloud, desktop, or inside physical medical device hardware, it retains its designation as medical device software.
Here are a few examples of medical device software:
- Pacemaker programming software (likely an accessory to pacemaker software, can be SiMD or SaMD)
- Insulin pump control software (typically SiMD or embedded software)
- Diagnostic imaging software (likely SaMD software using images from other medical devices)
- Electronic health record systems (SaMD software, depending on regulations, it might not be a medical device)
- Telemedicine software platforms (probably SaMD running on generic hardware platforms)
Standards can be helpful to meet otherwise challenging regulatory requirements. Choosing the right standards is vital to navigating regulatory approval processes.
The IEC 62304 standard, a globally recognised standard for medical device software, is particularly important.
The IEC 62304 standard applies to:
- Medical devices with embedded software, and
- Standalone software — known as Software as a Medical Device (SaMD).
The evolution of medical device software development: The role of IEC 62304
In the early 2000s, it became clear that the medical device industry needed a standard to ensure the safety and effectiveness of medical device software. Software was becoming an essential component of a wide range of medical equipment during this time, from basic monitoring devices to complex diagnostic and therapeutic machines.
An infamous example is the Therac-25 Radiation Therapy Machine.
In the Therac-25 case, a main issue was identified within its control software, which was riddled with numerous glitches. One critical glitch was a ‘race condition’ that happened when the timing or sequence of operations led to a hazardous situation.
If an operator entered treatment information quickly and the therapy followed immediately, the software could wrongly instruct the machine to release a strong radiation beam without having the correct target in place. This could result in a patient’s exposure to significantly high radiation levels, resulting in severe and sometimes fatal injuries, such as:
- Burns
- Tissue damage
- Organ dysfunction
While it’s uncertain if implementing IEC 62304 would have prevented all the issues with Therac-25, the aim is to prevent this type of adverse events.
The IEC 62304 standard includes essential steps like conducting software risk management and performing comprehensive software system testing.
Similar to a travel checklist that ensures you don’t forget to pack your toothbrush (again!), compliance with the IEC 62304 standard provides a systematic approach that can significantly minimise the probability of glitches causing harm to people.
And, just like a travel checklist proves most beneficial before and during your journey rather than upon your return, the same applies to the IEC 62304 standard; its main focus is to help throughout the development process.
The main processes in IEC 62304
IEC 62304 is organised into five main processes with underlying requirements:

Many read the standard as a predetermined order of activities. But it shouldn’t be understood that way. The standard’s introduction specifies the following:
“This standard does not prescribe a specific life cycle model. The users of this standard are responsible for selecting a life cycle model for the software project and for mapping the processes, activities, and tasks in this standard onto that model.”
If you’re want to use Agile methodologies, you can do so and meet the requirements of IEC 62304 as long as you can justify your chosen technique and correctly fulfil the standard’s requirements.
Medical device software developers are often confused about the requirements relating to documentation. IEC 62304 requires documentation, but it doesn’t prescribe a specific format. You can decide on how you want to create documentation and records that provide evidence of activities being carried out.
For instance, if you already operate with a reliable ticketing system, it might be perfectly adequate to demonstrate the traceability between software unit design and unit testing.
Similarly, if you have a software-based requirements management system, you may not have to create large amounts of paper documents.
In the IEC 62304 introduction, you find the following:
”This standard does not prescribe the name, format, or explicit content of the documentation to be produced. This standard requires documentation of TASKS, but the decision of how to package this documentation is left to the user of the standard.”
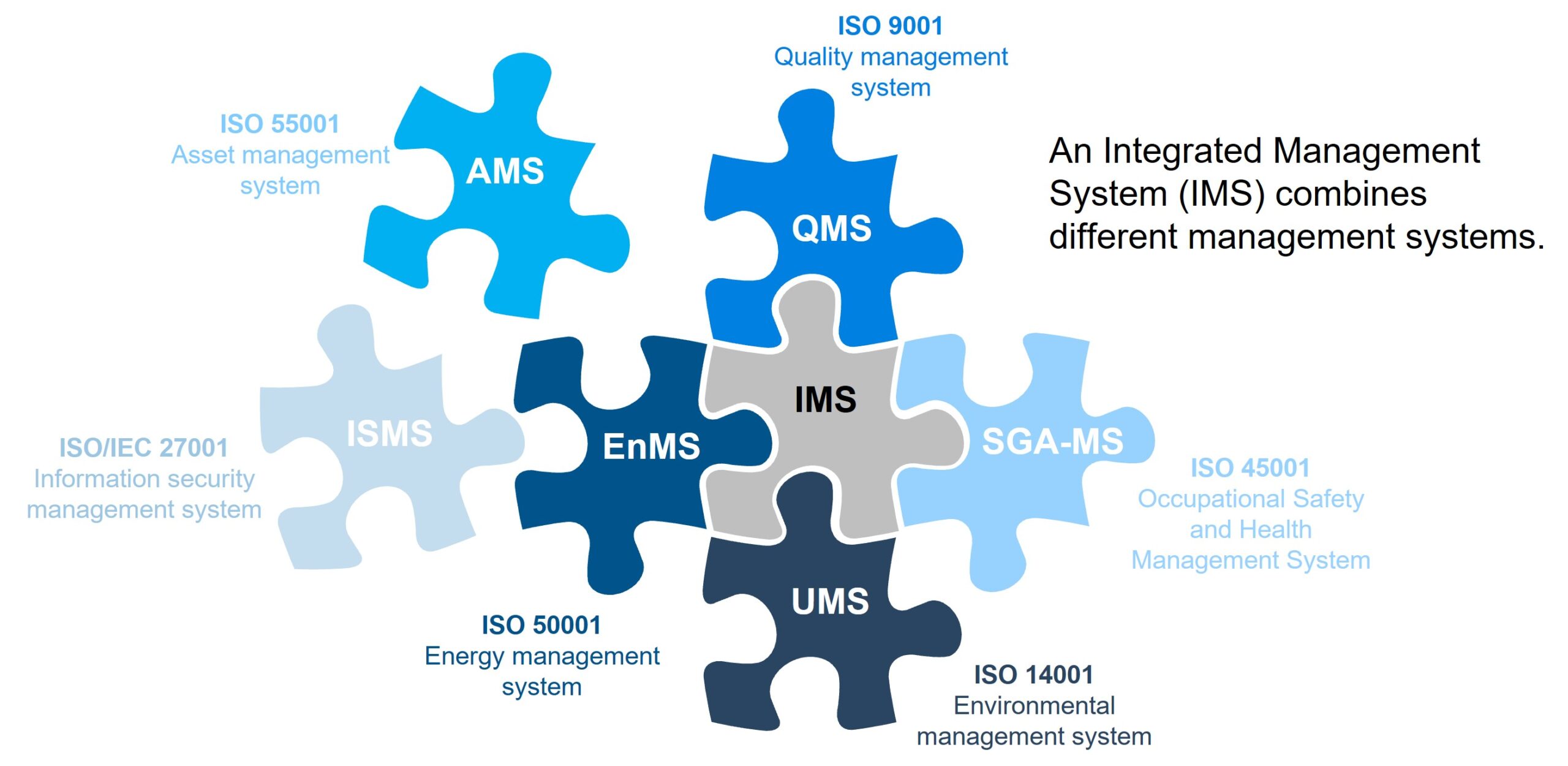
Understanding the relationship between IEC 62304 and other standards
There are many applicable standards for most medical device products, and a lot of them will also apply to medical device software. IEC 62304 is intended to coexist in an ecosystem of other standards. For example:
- ISO 14971 on risk management
- ISO 13485 on quality management systems
- IEC 62366-1 on usability engineering
- IEC 82304-1 a product standard for health software (applicable to SaMD)
- IEC 60601-1 on electrical safety (patient)
- IEC 61010-1 on electrical safety (equipment)
Medical device software and risk management
The most crucial standard to implement alongside IEC 62304 for medical device software is the ISO 14971 on risk management. It is a prerequisite for IEC 62304 to work as intended (aka normative reference).
Having a quality management system (QMS) is a requirement of the IEC 62304. The most common standard used to establish the QMS would be the ISO 13485.
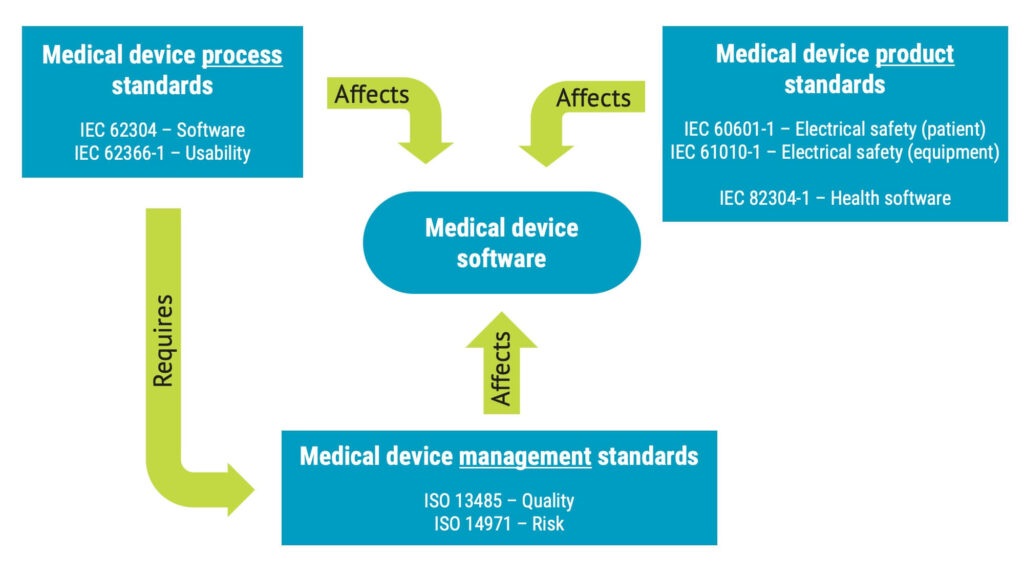
What is software of unknown provenance (SOUP)?
Software of unknown provenance, (SOUP) is software component that is not developed for use in medical devices but that you want to include in a medical device software system.
This could include software such as:
- Operating systems
- Databases
- Libraries
SOUP can originate from different sources, such as open-source projects, a company delivering generic software components or, eventually, a source code snippet from another project in your organisation.
Using SOUP in medical devices requires risk analysis and a robust software design. The purpose is to ensure that the reliability of the SOUP components won’t compromise the medical devices’ overall safety, performance, and efficacy.
In the US, the FDA term Off-The-Shelf (OTS) is commonly used. The definitions are similar but not identical.
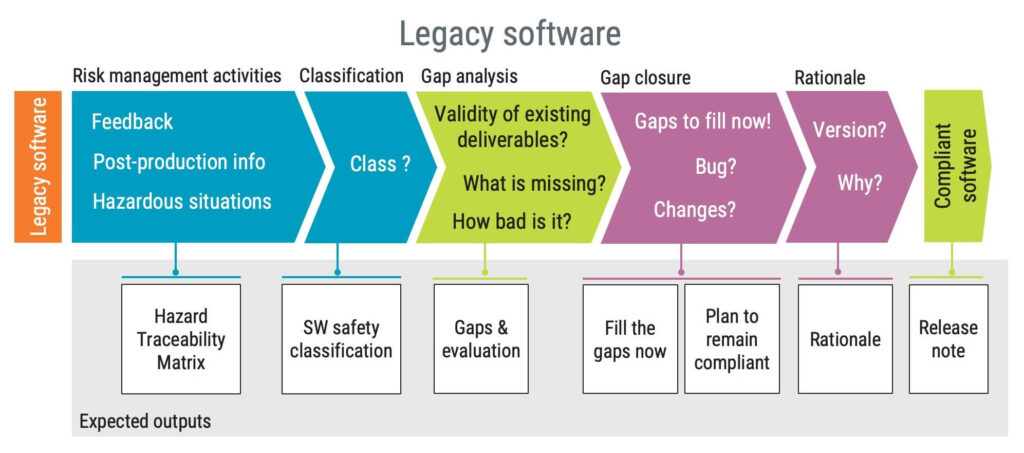
Legacy software
In contrast to SOUP components, legacy software refers to a medical software system that has been legally placed on the market and continues to be sold today. However, it lacks the necessary objective proof that it has been developed in line with the current version of this standard.
There are three scenarios where the concept of legacy software can be used:
- The software was developed as a medical device but before the current version of the standard.
- New functionality pushes an existing, non-medical device software into the medical device domain.
- Regulations become more stringent and broaden the scope of medical devices.
If you want to bring legacy software into compliance with IEC 62304, here is the required process:
Security
Security, or cybersecurity, refers to the measures and practices taken to protect computer systems, networks, and data from unauthorized access, attacks, damage, or any form of disruption.
Impact assessments of security risks are often done against CIA, which is NOT the Central Intelligence Agency from the US, but:
- Confidentiality – how to protect sensitive information, for example patient data.
- Integrity – how to ensure a device is safe to use in case of a security breach.
- Availability – how to ensure continued services delivery in case of a hacker attack.
IEC 62304 doesn’t say much about security. Still, you are supposed to identify and include security requirements in the software requirements.
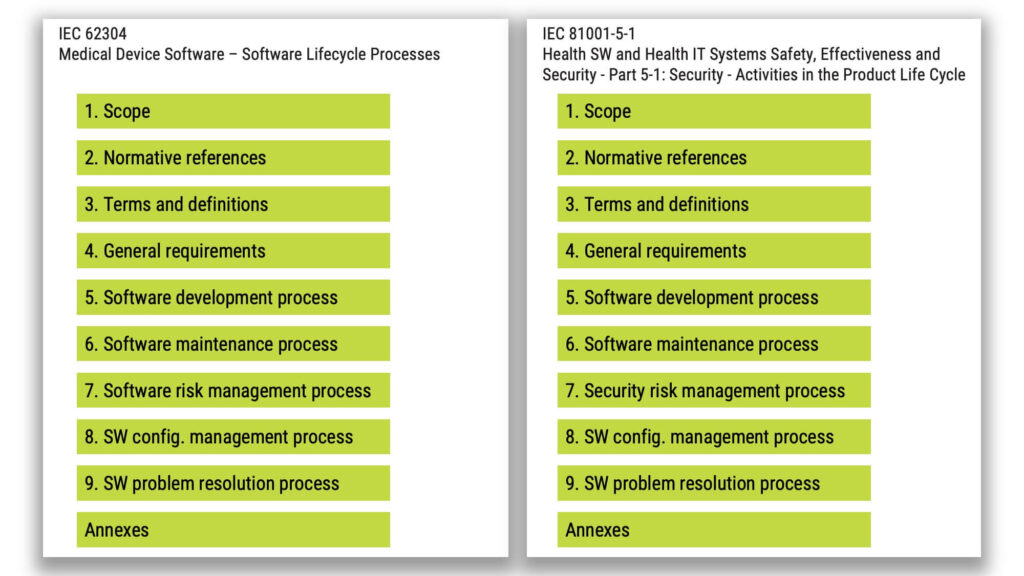
To support the identification of security requirements, you are recommended to take a closer look at the standard IEC 81001-5-1, which is structured like IEC 62304, making it a perfect add-on standard.
Software system, items, and units
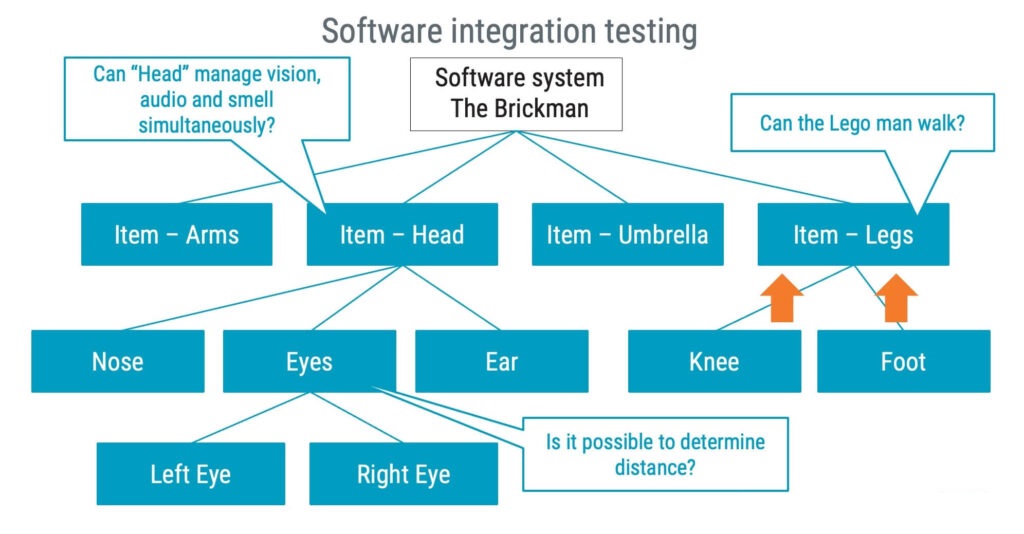
A ’software system’ is a combination of one or more software items that function together for a specific purpose or to achieve a specific result. In simpler terms, the totality of software parts contributes to a medical device’s functionality. ‘Software items’ are essentially subsets of the software system. They are the individual building blocks that combine to form the software system. Each software item has a specific role in the system, much like “The Brickman” pieces below coming together to form a complete character.
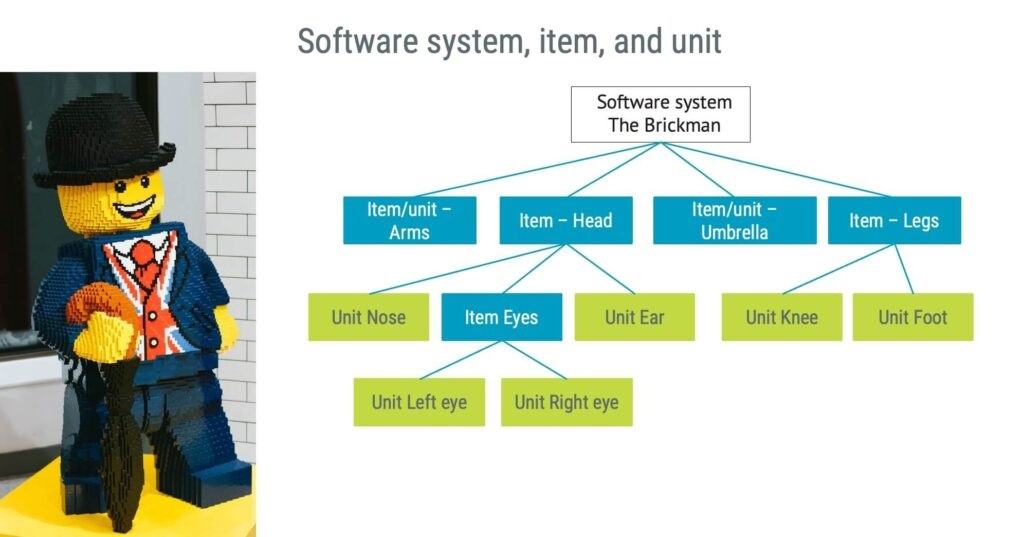
Finally, ‘software units’ are even smaller, more granular components than software items. Software units represent the smallest testable parts of the software – the basic building blocks that make up the larger pieces (items) of ”The Brickman”.
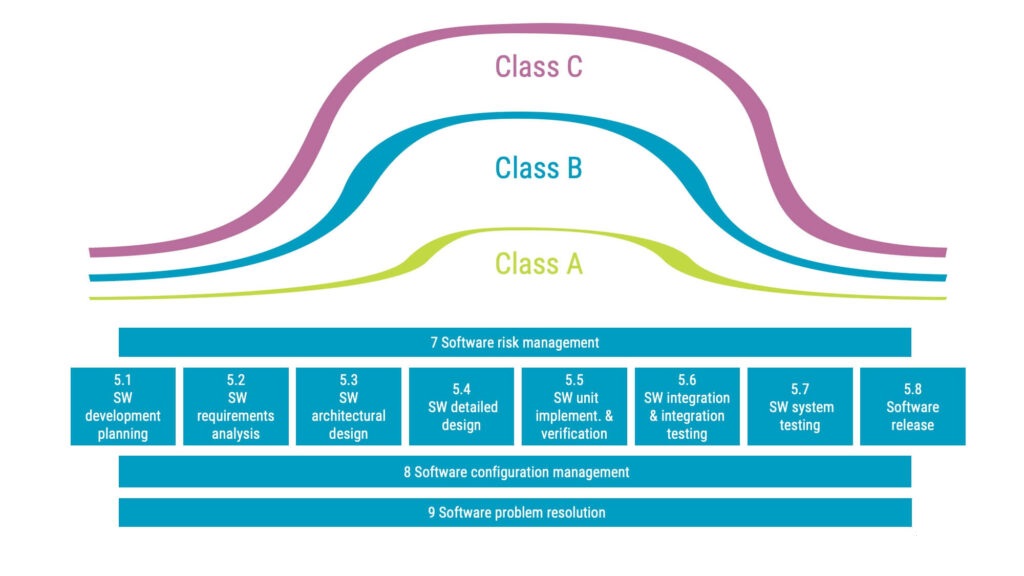
Delving into the IEC 62304 software safety classification
The main aim of the standard is to lower risk. The more potential risk the software can cause, the stricter are the requirements of the standard.
In the standard, this is referred to as ‘software safety classification’. The classification is split into classes A, B, and C with class C used for software with the highest risk profile.
In essence, the principle is straightforward: The higher the risk, the more you are expected to do. A few examples to make it more tangible:
- Unit verification is required for class B and C while there are no such requirements for class A software.
- Detailed design documentation is required for class C but not for class A and B.
The software safety classification is based on the potential injury the software can contribute to and can be summarised as follows:
- Class A: No injury
- Class B: Non-serious injury
- Class C: Serious injury or death
The formal definition of serious injury is:
“injury or illness that:
a) is life-threatening,
b) results in permanent impairment of a body function or permanent damage to a body structure, or
c) necessitates medical or surgical intervention to prevent permanent impairment of a body function or permanent damage to a body structure”3.33, IEC 62304
The standard provides a flowchart guiding the classification work. A somewhat simplified diagram for this is used below, but the content is the same.
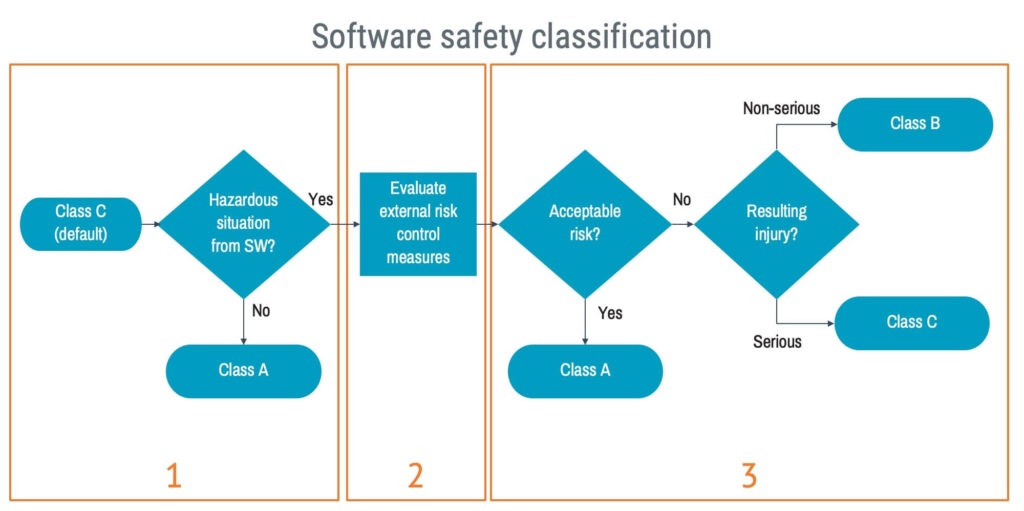
There are three essential steps to consider in the classification process.
1. Identify hazardous situations
Starting with your system risk analysis, identify hazardous situations that could arise from software failure. If you don’t find any, congratulations! Your software is a class A software.
2. Evaluate the effectiveness of risk control measures external to the software
Identify external risk control measures for each hazardous situation and evaluate their effectiveness. If they are good, they can offload the safety burden from the software, and that particular hazardous situation can be excluded from the software safety classification.
3. Determine what is acceptable
Based on your evaluation in step two, evaluate the acceptability of the risks related to the software. If the risks are acceptable, your software is a class A software.
If there are unacceptable risks, assess the potential injury resulting from those risks to determine whether class B suffice or class C is required.
Software safety classification – common mistakes and misunderstandings
Software safety classification does not determine how safe your product is
The classification of software safety dictates the rigour of the process, as the assumption is that enhanced examination of development tasks will improve safety.
However, the standard leaves the determination of product requirements and risk control measures to the discretion of individual manufacturers.
The benefits of the software cannot be used to lower the software safety classification
When classifying risks, it’s important to remember that it’s not about comparing the benefits of having the software versus not having it. This method is known as benefit-risk analysis and is derived from the ISO 14971 risk management approach. This approach is only used when further risk controls aren’t feasible. Thus, you cannot reduce the software safety classification because of the degree to which the software benefits its final users and patients.
Implementing risk control measures in the software itself does not lower the classification
Unfortunately, software can always fail. As a result, simply incorporating risk control measures within the software itself does not lower the safety classification. This does not mean that you shouldn’t implement risk controls in the software to decrease the probability of software failures, it only means you can’t lower the software safety classification as a result of risk controls within the software itself.
Getting the probability of software failure wrong
ISO 14971 talks about the probability of occurrence of harm, Po. However, Po can be split into two parts: P1 and P2.
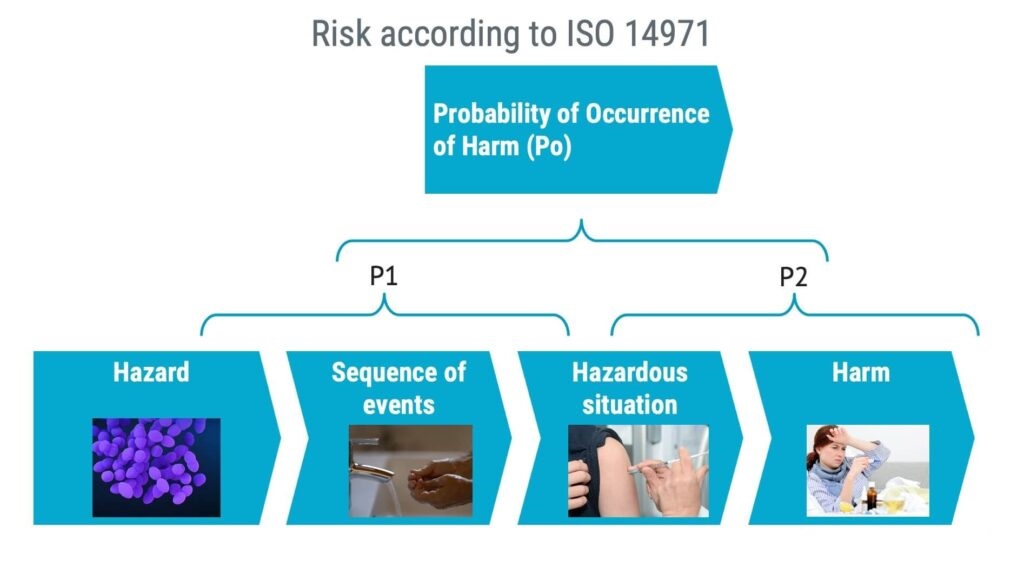
When it comes to software safety classification, you should assume that the software could fail at any time, which means that when estimating the probability of software failure, you should set it to 100%.

Even if software failure is the sole part of P1, the probability of the hazardous situation leading to harm (P2) may still be very low resulting in a low Po.
In summary, getting the concept of P1 and P2 right can have a significant impact on assigning the software safety classification.
Risk management in the IEC 62304 standard
Medical device software risk management should be performed according to both IEC 62304 and the risk management standard ISO 14971. While ISO 14971 focuses on the overall process and identifies hazardous situations, IEC 62304 works bottom-up to identify how the software can fail and contribute to hazardous situations.
So, continuous iterations will occur between the software development process and the system level above throughout the software life cycle.
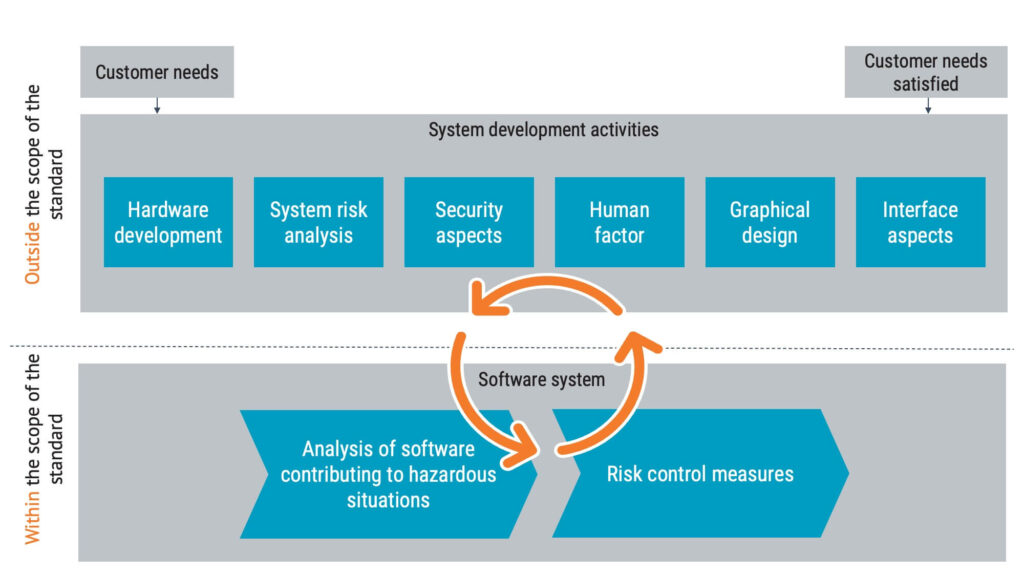
Software risk control measures
In the context of IEC 62304, software risk control measures aim to reduce the probability of the software failing. There are three main areas you can explore in identifying suitable risk control measures within the software system:
1. Development process
Following a strict procedure is believed to reduce the probability of failures. This can, for instance, be peer reviews and the use of static code review tools. Or why not use a good coding guideline?
2. Software system
You can work with architectural design decisions to reduce risk at the system level. For instance, using an operating system to prioritise critical tasks can be seen as a risk control measure.
3. Software items/units
At this level, it gets close to good engineering practices such as implementing range checks and using checksums on critical data.
The sum of your risk control measures can be used to claim a reduction of P1.
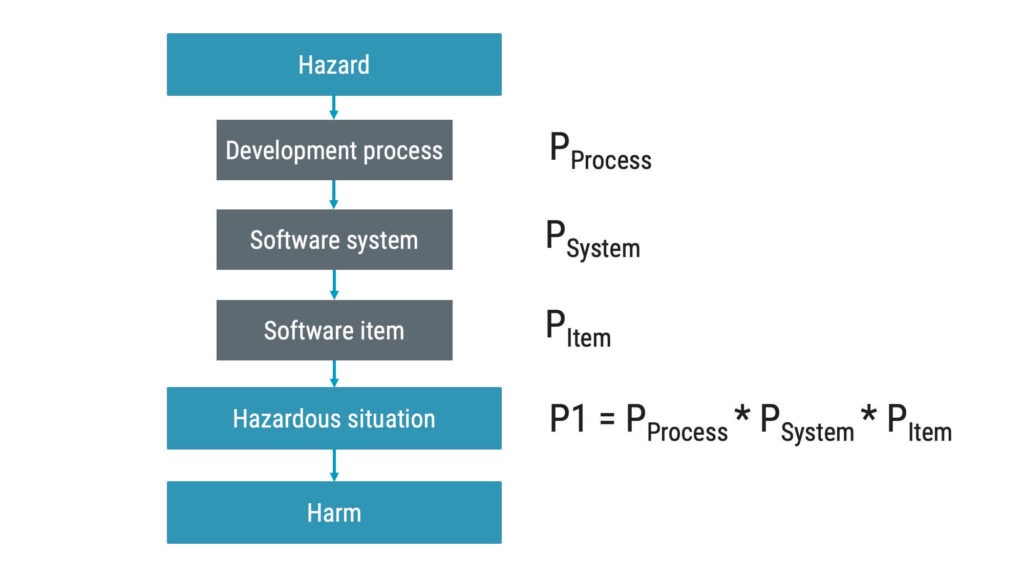
Assessing the efficacy of software risk control measures
Estimating the probability of software failure is nearly impossible. It is equally challenging to determine the effectiveness of software risk control measures. To address the challenges, the best practice is the following two steps:
1. Verify that the software risk control measure works as intended
Assume you have implemented a checksum feature. Provide evidence that the feature can detect incorrect data.
2. Work with relative probability
Without any risk controls, you should assume the probability of software failure is 100%. Now, assume you implement a risk control measure; how much is the probability lowered compared to without the risk control measure?
Let’s look at a scenario where you’ve implemented and tested a checksum. You might instinctively want to assume that the probability of failure has been reduced by 100%. However, don’t disregard the fact that an error could creep in during the checksum calculation. Therefore, a more realistic reduction of the probability of failure might be 99.9% rather than 100%.
Admittedly, this is an estimate, but when true statistical calculations aren’t feasible, an expert assessment based on good engineering judgment is an acceptable way forward.
Software causes
Software causes is the term used in IEC 62034 to identify why software can fail. Potential software causes are, in short, anything that can trigger the software to present undesirable behaviour – both inside and outside of the software.
Here are a few examples of causes from the technical report IEC/TR 80002-1, which is guidance on the application of ISO 14971 to medical device software:
- Alarms and alerts
- Critical power cycle states
- Critical user controls/Usability
- Display
- Hardware controls
- Monitoring
- Interfaces
- Data
- Diagnostic
- Security
- Performance
How do you document software risk management?
The software risk management trace you are expected to establish, following IEC 62304, shall demonstrate how software items, through causes, can contribute to hazardous situations.
A visualisation of the entire traceability chain is exemplified below:
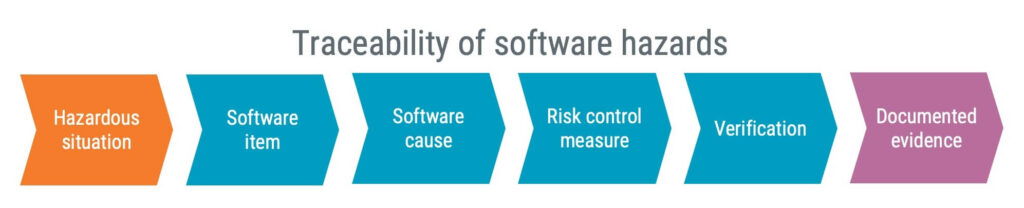
If you are a SaMD developer, most of your risks are likely to relate to the software. So, it might be convenient to expand your hazard traceability matrix (HTM) to include IEC 62304 specific information, such as ”software item” and ”software causes”.
If you’re a developer not specialising in SaMD, a more complex HTM could prove challenging. Therefore, your efforts may be better directed towards linking the system HTM and software risk management documentation via a hazard analysis.
This can be achieved by creating a table. This table would document linkage to hazardous situations and have columns denoting respective software items, causes, and appropriate risk control measures and verification strategies.

Software risk management – common mistakes and misunderstandings
Duplication of system hazard trace matrix
Software risk management according to IEC 62304 uses a ‘bottom-up’ approach to identify possible risks linked to the software. Your software risk management process should not duplicate system risk management work. For instance, identifying hazards and harm in two processes while it is sufficient to do so once in the system risk management process.
By avoiding duplicating activities, your software risk management work can be leaner and less burdensome.
Working with software risk management retrospectively – too late!
Unfortunately, it is common to find projects focusing on developing functionality with a ”wow factor” to impress project sponsors, while at the same time, neglecting risk management. Sometimes, risk management is performed as late as just before release to make regulators happy.
But remember, as medical device developers, our primary goal should be to ensure people are not harmed directly or indirectly by the software we develop. Risk management should not merely be about pleasing regulatory bodies. So, adding a couple of software risk control measures only to demonstrate compliance isn’t good enough!
Designing safe software starts from day one, making software risk management something that should be considered daily throughout the life cycle of the software.
IEC 62304 software development activities
It is commonly known in the software industry that a good design process is vital to quality, and there are no shortcuts. While this is a desired property for general-purpose software, it is a must-have for medical device software because people’s lives can depend on whether the software works or not.
“If you think good design is expensive, you should look at the cost of bad design.”Ralf Speth
To facilitate high-quality software, the IEC 62304 standard outlines the following activities that must be included in the software development process:
- Planning
- Requirements
- Architectural design
- Detailed design
- Implementation
- Integration and integration testing
- System testing
- Release
The concepts above are presented in a specific order in the standard, but besides planning, which has to be done at the start, you don’t have to finish one before starting the next.
For example, think about how requirements are managed in Agile compared to the waterfall approach. In Agile, you define the requirements for a limited number of functions and then work on implementing those requirements. In a more traditional approach, you aim to define all the requirements up-front. Either way, the end result should be the same: a set of requirements and a matching software implementation.
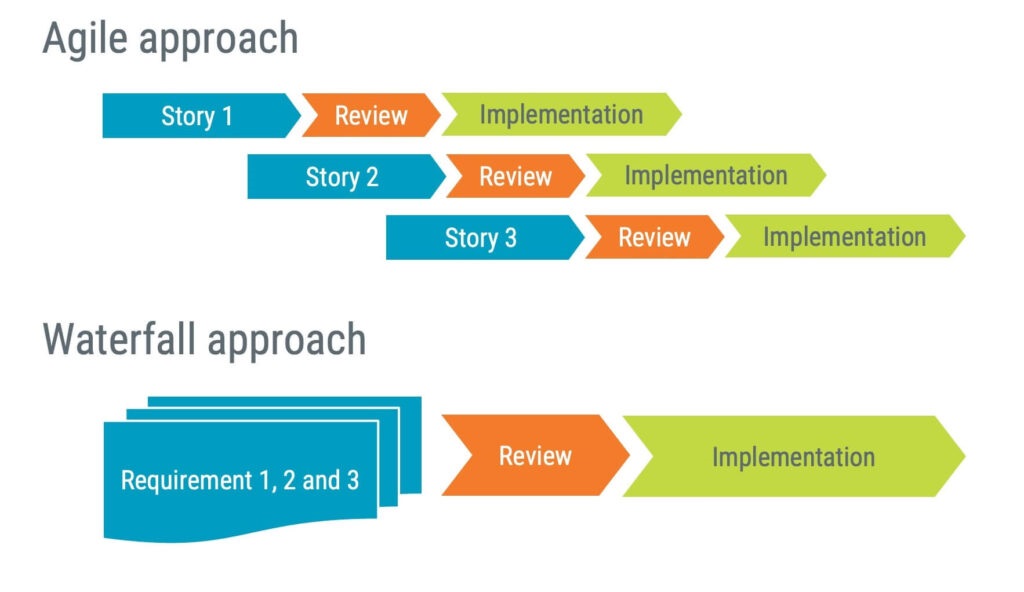
How to plan medical device software development
Planning is about reducing the likelihood of undesirable surprises and guiding the development work.
“Success depends upon previous preparation, and without such preparation, there is sure to be failure.”Confucius
The software safety classification determines the rigour of of the software development process, and therefore also the level of detail in the software planning.
The left-hand side of the high-level figure below shows the areas you should have in place before starting the development work.

Processes
Which methods are you planning to use? You can reference existing processes in your quality management system or set out a preferred process in the plan. Explain how your approach aligns with the standard’s criteria, whether you favour XP, Scrum, Kanban, test-driven development, or another process.
Deliverables
What and when will you create outputs from the project? This is not only about the software binary but should also include the documentation that must be established.
Traceability
Traceability is required to demonstrate that software is doing what it is intended to do. This is achieved by demonstrating how software requirements are linked to a source, how they can be identified in the design, and how requirements are tested.
Remember to include traceability between software risk control measures, hazardous situations, and applicable risk control measures.
Configuration and change management
How is version numbering expected to work? Consider if you want to split the software into multiple components with individual version numbering. A multiple-component approach might be beneficial when determining regression testing in response to changes because changes implemented in one component might not impact other software parts if properly designed.
Speaking of changes, the plan shall address how changes shall be managed. For some changes, like bugs during development, you can likely leverage an existing ticket system, while changes after release might need a more detailed review before allowing changes.
Problem resolution
Problems, aka bugs or anomalies, will be part of your development. The plan shall address how problems should be reviewed to ascertain if they should be corrected and if so, what actions must be taken.
A problem-resolution process can often be managed within an existing ticket system, assuming you include the details required by IEC 62304, for instance, if there is a need to advise parties about the problem.
Software requirements specification in IEC 62304
Software requirements define your product and should provide a definitive description of the specific functions, performance, interface, design constraints, and inherent characteristics that a software product must have. This, of course, also includes security-related requirements and risk control measures. Getting the requirements right is crucial for the development work, and requirements engineering for medical devices is an important knowledge area.
The quality of the requirements is not only important for the for the development work, but also to ensure that maintenance can be carried out without jeopardising the safety of the software and medical device. Consequently, maintaining software requirements is a life cycle activity.
An essential IEC 62304 activity is to verify software requirements (clause 5.2.6). In this context, ”verify” shall not be confused with ”testing”; this activity is about reviewing the quality of the requirements. In short, the requirements shall be:
- Unambiguous
- Implement system requirements and risk controls
- Identifiable
- Testable
- Traceable
The software development plan shall describe how requirement traceability is established. A good practice is to draw a flowchart, as shown in the example below. A visual traceability flowchart can also be invaluable during audits and technical file reviews.
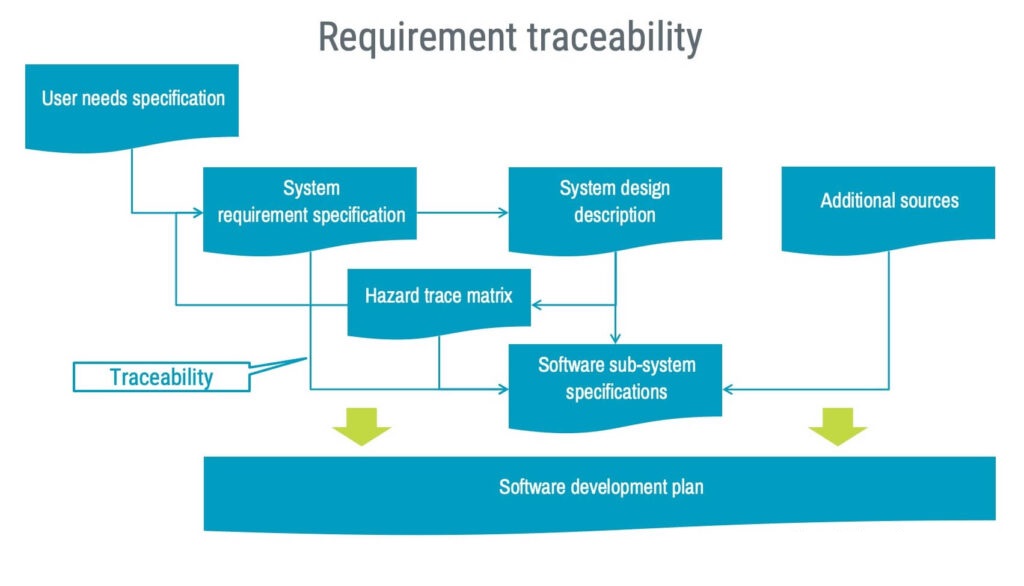
Architectural design
Putting effort into a solid software architectural design is essential. It lays the groundwork for the next steps and can influence future maintenance. IEC 62304 recognises certain critical activities in this process:
Transform requirements into an architecture
Use best engineering practices! How many items make sense? Should functionality subject to frequent changes perhaps be gathered in a single item?
Develop architecture for the interfaces of software items
Consider whether you can utilise established interfaces, perhaps within an operating system. At times, it may be enough to use standard function calls. However, there could also be instances where you require a more sophisticated mechanism to establish interaction between items.
Specify requirements for SOUP items
If you decide to use SOUP software, you should define what you expect from using it. Equally important is to identify if the SOUP items have any prerequisites to function correctly. Together, this information is used to test the SOUP and demonstrate that the SOUP fits your software’s purpose.
Identify segregation necessary for risk control
Even though this is a class C requirement only, it may still be worth considering for all classes. If the architecture can isolate critical functionality to a few items, perhaps that enables you to down-classify class B (or C) items to lower classes, easing the development work.
Whilst there is no one-size fits all blueprint for documenting software architecture, diagrams, such as the ones below, are commonly used.
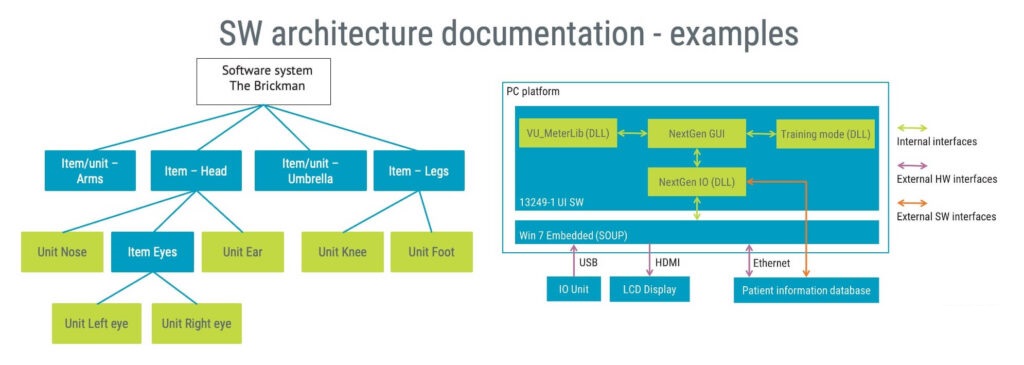
Should your design extensively involve interactions amongst items and other software systems, you can consider a sequence diagram.
Software detailed design
The detailed design is about refining the software architecture until it is represented by software units.
This is one of the activities in IEC 62304 that differentiates class C the most from the other classes by defining requirements on:
- Detailed design for each unit
- Detailed design for interfaces
- Verification of detailed design
The standard is vague on the expected evidence of these activities. Still, the purpose is to promote design work and reflection before the coding work starts to minimise the risk of ad-hoc coding.
How should detailed design be documented? The main goal is to create documents that aid the implementation work. The style in which this is accomplished may vary, but do remember that a detailed design could be required for regulatory submissions. Therefore, it’s smart to consider how you can present the detailed design in printable format, such as a PDF-file.
Here are a couple of examples to consider:
Write the software detailed description documents
Documents can be traditional Word documents but could also be captured in a wiki.
Capture design information in a ticket system
If you are working with user stories and tasks, the design process can be documented in relevant tickets.
Use the source code
Using a structured approach to commenting, necessary design documentation can be kept in the source files. The starting point can be an empty skeleton describing the detailed design, which is then populated with source code in the implementation phase, as in the example below.
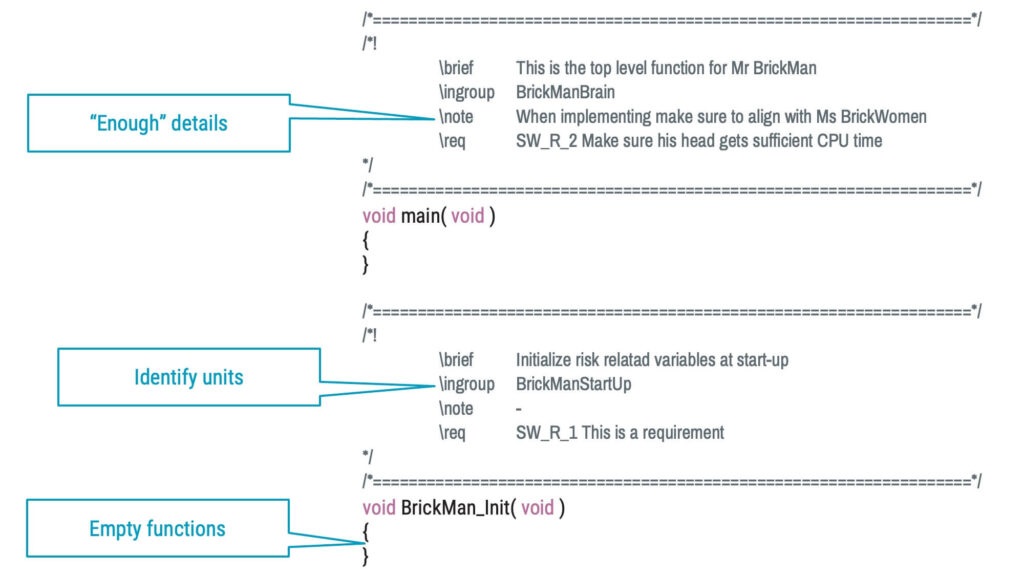
Software implementation
To a lot of programmers, this is where the fun starts – the actual coding work!
In addition to coding, it is essential to remember unit verification. In many cases, unit verification can be performed with the help of a unit test framework. However, unit testing isn’t the only viable option; you can establish any strategy, method, or procedure you find adequate. This can, for instance, include:
- Peer reviews
- Static code analysis tools
- Conformance with coding standards
Often, you will see a mix of approaches employed to form a solid strategy for unit verification and testing.
Integration and integration testing
Integration is about assembling software units into items and items into a software system. When verifying successful integration, you confirm that all units and items can be assembled without errors.
You might think about combining integration testing with system testing. However, it’s worth noting the advantages of keeping these as separate tasks, because finding interface and interaction issues among integrated modules can be tough during system-level testing.
Returning to ‘The Brickman’, it might be beneficial to conduct integration testing on items to test their capabilities thoroughly before testing the entire Brickman.
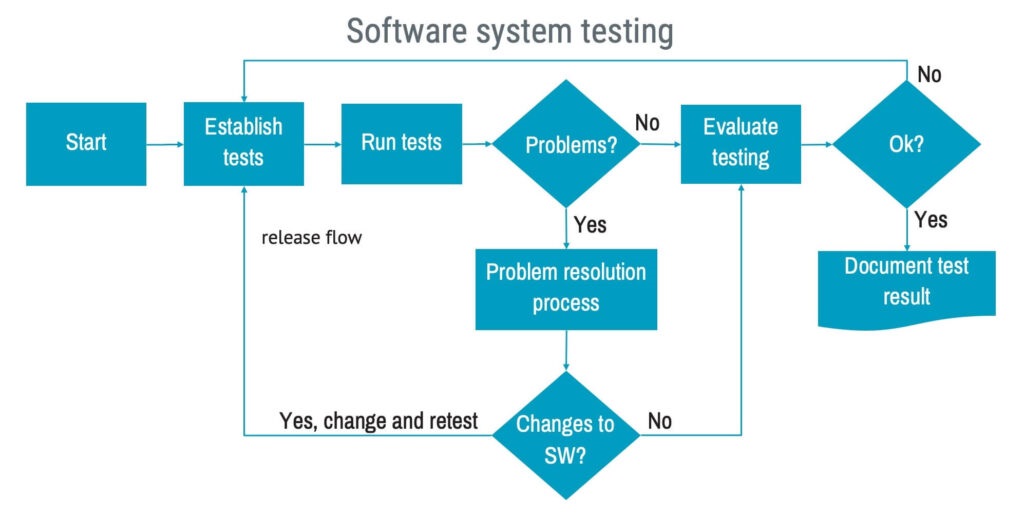
System testing
Software system testing is there to make sure the software works as it should and is safe to use. This can be complex, as tests may cover various ways the software is used and possibly on different hardware systems.
In clause 5.7.1 a) in the standard, this is expressed as:
“The manufacturer shall establish and perform a set of tests, expressed as input stimuli, expected outcomes, pass/fail criteria and procedures for conducting software system testing, such that all software requirements are covered.”
In IEC 62304, you’ll come across a collection of tasks that need to be performed during the system testing stage. To put it simply, these tasks form the following work process:
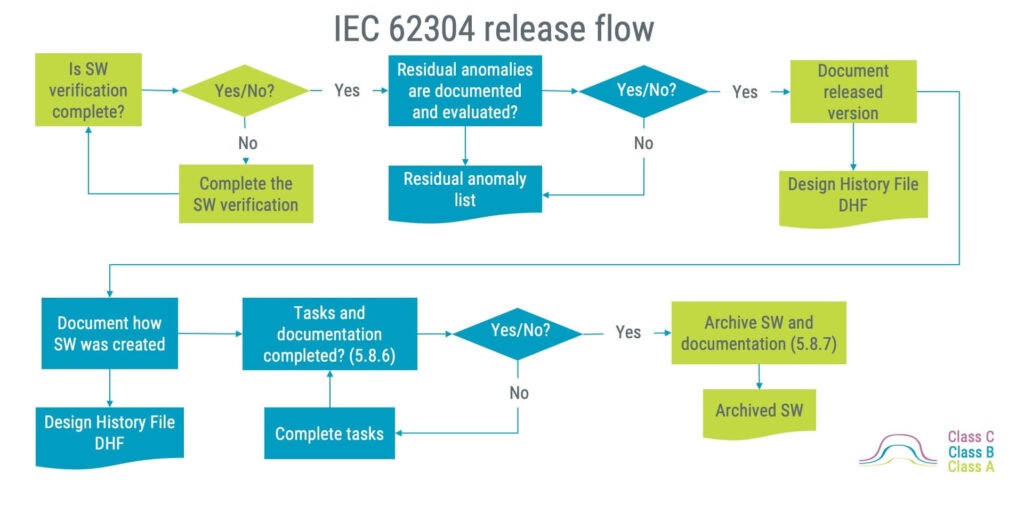
Software release
After all your hard design work, it is time for a release party. But, before the party begins, you have to assess if the software is release-ready, and if so, document what goes into the software release.
The flowchart below illustrates what must be done in an IEC 62304 software release. Worth noting is that it is acceptable to release software with ”residual anomalies” or bugs. This may sound awkward, but it may be acceptable if the bugs are evaluated as not contributing to hazardous situations or otherwise impacting the software functionality. Acceptable residual anomalies can for example be:
- Typos
- Misalignment in graphical elements
- Bugs with acceptable workarounds
How to maintain medical device software according to IEC 62304
The IEC 62304 standard acknowledges that after the software is released to market, its journey doesn’t end there. The software still requires regular monitoring, maintenance, and possibly updates.
The IEC 62304 standard requires the establishment of a maintenance plan. This plan should include steps for identifying problems, implementing solutions, and ensuring those solutions do not introduce new risks or adversely affect the software’s performance. This plan plays a significant role in the post-production life cycle of medical device software.
Remember, the key to successful maintenance lies in having a proactive approach, as opposed to a reactive one. It’s about identifying and resolving issues before they compromise patient safety or device performance.
In addition to dealing with identified problems, the maintenance plan should also address maintenance needs triggered by:
- Changes to SOUPs
SOUPs may be subject to upgrades, bug fixes, patches and obsolescence.
- Cyber security updates
For products exposed to security threats, there is likely a continuous flow of information that must be evaluated.
A good practice is to take a risk-based approach to how frequently SOUPs and cyber security information shall be evaluated and defined in the software maintenance plan.
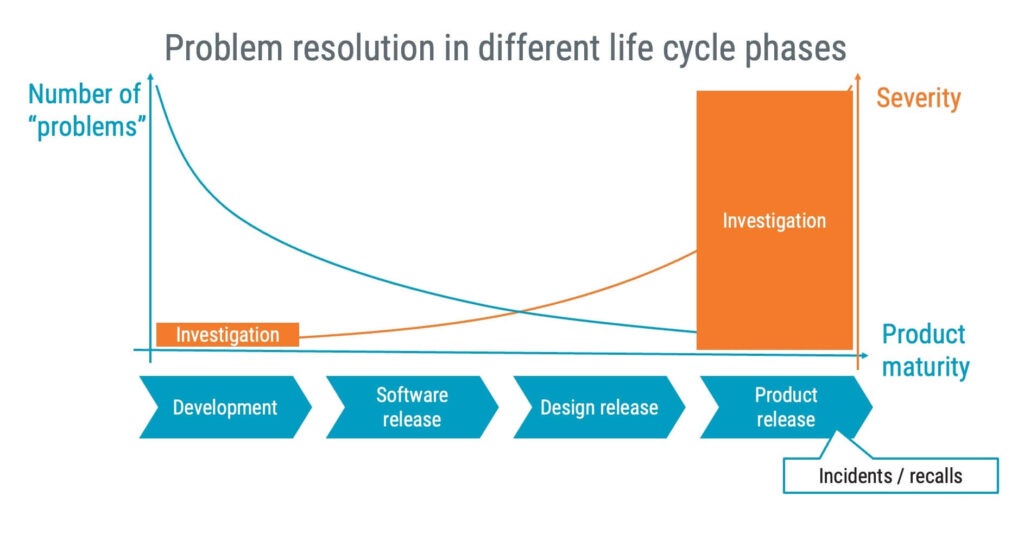
Problem resolution
Unexpected issues are part and parcel of developing medical device software. The IEC 62304 standard provides the roadmap for approaching problem resolution when such situations arise. A systematic and structured approach towards addressing these issues increases efficiency and reduces the potential for new errors in your software as a result of fixing existing errors.
Fundamentally, problem resolution under IEC 62304 involves identifying, analysing, and correcting anomalies. An anomaly can represent anything from a code bug to a design flaw. It’s crucial to document these anomalies meticulously, offering clear descriptions and circumstances under which they occur.
When setting up a problem-resolution process for software, it’s wise to start with a light-weight problem resolution process to avoid overwhelming the early stages of development with administrative work relating to problems.
In the early phases, you might find that a basic ticketing system is enough to track your progress. However, as your project advances, a more sophisticated approach is recommended to perform more in-depth investigations that can prevent future recalls of the medical device.
Change control
Controlling changes is required in IEC 62304. It’s not just tech talk but a fundamental principle to ensure the quality and security of your medical device software. So, what does controlling software changes comprise?
Change control is all about managing any changes to your software in an organised way. To see it more clearly, imagine your software as a large, busy city. Every software unit is like a building in the city. Just like city planners, you wouldn’t randomly destroy or build buildings.
Instead, a structured process involves practicality studies, analyses, and regulatory procedures. Similarly, in software development, change control ensures that changes are necessary and beneficial and won’t disrupt the overall functionality and safety of the software.
When designing a change control process, a balance should be struck between being in control and efficiency of the process. An overwhelmingly rigid process can effectively kill a development team’s performance without any clear benefits.
Two common mistakes are:
1. Too much documentation too early
If a software engineer encounters a minor bug, in most cases, it should suffice to document the finding and correct the issue. Since there are no changes to the requirements, it is a correction of what has already been approved, and the value of triggering a significant investigation with corresponding documentation is in most situations not necessary.
2. Involving too many in the approval process
In some organisations, there is a tendency to invite the entire company into the change control process. But less is more! Question what value individuals bring before they are invited to take part in the process.
The image below is not an IEC 62304 requirement but is intended to provide food for thought on how to design a change control procedure with flexible rigour while still meeting the requirements of IEC 62304.
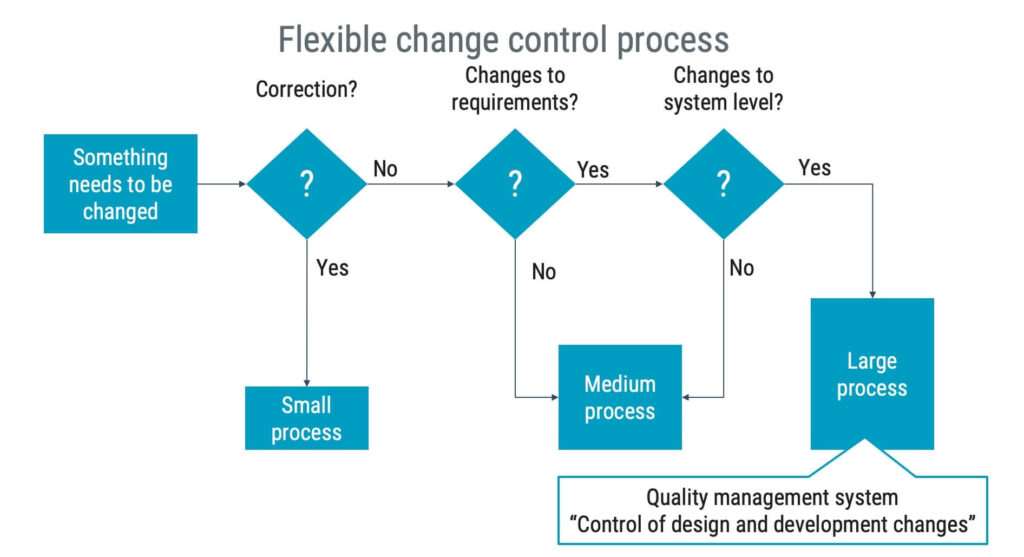
How to implement the IEC 62304 standard successfully
Understanding Agile in the context of IEC 62304
Agile practices can be pivotal in efficiently implementing IEC 62304 in your medical device software development. With Agile methodology, you focus on iterative, incremental deliverables, emphasising transparency, flexibility, collaboration, and adapting to change.
This iterative approach aligns perfectly with the requirements of IEC 62304:
1. Excellent for software risk management
Agile facilitates the consistent application of a risk management process that is required by IEC 62304. It encourages early and continuous risk identification and mitigation throughout the software life cycle, which is a requirement of IEC 62304.
2. Promotes clear requirements
Agile promotes frequent communication and feedback loops, ensuring clarity in development stages and meeting most verification requirements as defined by IEC 62304. (Assuming you keep records of what you are doing.)
3. Early detection of design flaws
Agile practices drive continuous integration and testing, an essential requirement for software system testing in IEC 62304.
4. Integral change management
Agile’s responsiveness to change aligns with IEC 62304, allowing you to manage changes to software requirements and system design actively, ensuring traceability and control.
However, remember that while Agile offers substantial benefits, it’s your responsibility to tailor it to suit the regulatory constraints of your industry.
Are you an adopter of Agile methodologies? AAMI’s technical report TIR 45 offers valuable insights on effective medical device software creation procedures. It’s an outstanding resource for those aiming to perfect their development practices.
Look at the complete picture
The software development will often have different stages and phases compared to other areas of development. When the processes don’t mesh well, it can result in sluggish organisational performance. In the journey to implement IEC 62304, it’s helpful to take a holistic view of the entire process from start to finish.
Consider, for example, simulating updating the technical file for the following product release and ask yourself:
- How easy is it to identify relevant deliverables from the software development process?
- Do you find evidence that risk control measures have been implemented in software and tested?
- Can you easily establish a traceability matrix with high-level requirements traceable to software requirements and relevant verification evidence?
Decide how you want to document
IEC 62304 has numerous requirements that necessitate the creation of documents or records. However, these documents do not always need to be physical and may not require signatures. It is sufficient to ensure that the relevant information can be identified and is under version control. This can be achieved by utilising an existing configuration management tool that is used to version control software.
Let’s look at unit testing as an example:
All information, including acceptance criteria and test results, can be kept close to the source code and version controlled with the source code.
The best practice in this approach is to format the unit test documentation to make it presentable and, if needed, exportable to a PDF file.
What are the consequences of not adhering to the IEC 62304 standard?
While conforming with the IEC 62304 isn’t a mandatory regulatory requirement, it is an expectation from more or less all regulatory authorities and notified bodies. If you do not conform to the standard, you should be able to explain why, which is easier said than done.
The IEC 62304 plays an important role in satisfying regulatory requirements and eases the facilitates the approval process of medical devices. As an illustration, the recent FDA guidelines for the pre-market submission of software-inclusive medical devices recognise the IEC 62304.
Thus, by declaring conformity with this standard, submission documentation requirements may be significantly lessened, making the whole process more streamlined and manageable.
Challenges when implementing the IEC 62304
Implementing a software development process according to the requirements of IEC 62304 can be challenging. Some organisations will struggle with:
- Implementing the IEC 62304 without causing unnecessary administrative work which could slow down the development process and reduce organisational efficiency.
- Combining IEC 62304 conforming software with Agile development.
- Making all programmers embrace a process that helps achieve high-quality software.
The challenges above are addresses by the Introduction to software for medical devices and IEC 62304 and Introduction to SaMD, IEC 62304 and IEC 82304-1 courses. You can also reach out to us for a custom in-house course tailored to your needs.
Would you like to learn more about Medical Software Development?
With our medical device software course selection, you can choose between Software for Medical devices and IEC 62304 and SaMD, IEC 62304 and IEC 82304-1 depending on your interest and need.
The courses are suitable for anyone working with software development, such as R&D engineers, quality assurance department and auditors of software development. The courses do not cover actual coding.
Or if you’re looking for a tailored training to align with your company’s specific needs – contact us for inhouse training options.