Risk management for medical devices refers to the process of ensuring that medical devices are safe. Performing risk management is a regulatory requirement in all major markets for medical devices.
The go-to standard for risk management is the ISO 14971 – Medical devices application of risk management to medical devices, which includes requirements for managing risk from the entire life cycle of a medical device, from initial conception until final decommissioning.
This illustrated guide was written by Peter Sebelius, a member of the technical committee (TC210), who authored the ISO 14971:2019.
This guide will:
- provide a useful overview of risk management for medical devices and ISO 14971; and
- address common misconceptions within risk management.
What is risk?
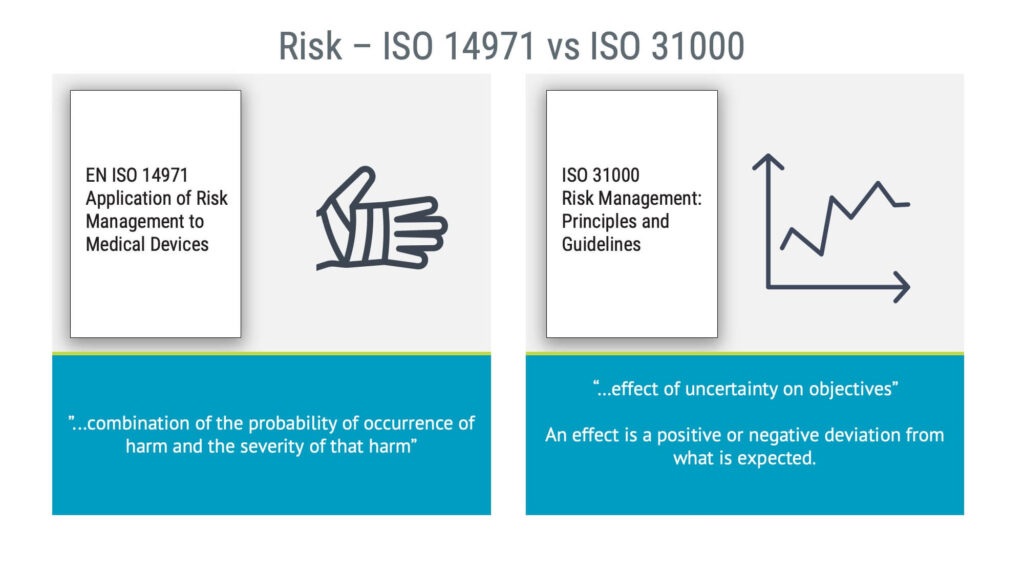
For medical device risk management, ISO 14971 is the go-to standard. ISO 31000 is another risk management standard from ISO. There is a fundamental difference between ISO 14971 and the ISO 31000 standards: their definitions of risk. The ISO 14971 definition of risk is about product safety and is concerned with harm to people.
The ISO 31000 standard has a broader definition of risk, as it comprises any effect on objectives, whatever it may be. Not only that, it also addresses positive risks, or opportunities, to use another word. Performing corporate or project risk management is always a good idea, but it cannot replace product safety risk management.
Risk management for medical devices is about product safety; it is not about:
- corporate risk;
- financial risk; or
- project risk.
Therefore, avoid mixing ISO 14971 risk management with ISO 31000 risk management.
The definition of risk in ISO 14971 is:
“The combination of the probability of occurrence of harm and the severity of that harm.”
What is risk management for medical devices?
Risk management for medical devices helps manufacturers identify potential hazards and assess the associated risks to take steps to reduce those risks.
The formal definition of risk management according to ISO 14971 is:
”The systematic application of management policies, procedures and practices to the tasks of analysing, evaluating, controlling and monitoring risk.”
The definition from ISO 14971 is, of course, technically correct but hard to remember. A more straightforward way of expressing what risk management is would be to say that it is:
“The systematic and continuous work to reduce risk.”
“Systematic” because if you work systematically, the risk will be reduced, and you will likely reach most of the goals you have set.
“Continuous” because risk management does not end when you have developed your product. The process should go on until your product is no longer used.
“Reduce risk” because this is what risk management is all about: reducing risk.
The goal of risk management is to create safe products. Many people define “safe” or “safety” as “free from risk,” but no medical device is entirely free from risk. There are risks associated with everything.
In ISO 14971 “safety” is defined as:
“Freedom from unacceptable risk”

Freedom from unacceptable risk is a fundamental principle of risk management for medical devices. It means the risks associated with a medical device should be reduced to an acceptable level.
The definition of what constitutes acceptable risk for a medical device should be decided by top management and be based on, among other things, regulatory requirements.
Why is risk management important?
Avoiding paying damages
Effective risk management in developing and manufacturing medical devices is paramount to mitigate some of the risk of having to pay damages arising from product liability cases.
From this point of view, the investment in risk management can be seen as a financial decision to reduce the expected future costs of paying damages.
Regulatory requirements on medical device risk management
Risk management is a regulatory requirement. Without performing risk management and meeting the requirements of ISO 14971, the doors to most major medical device markets worldwide, including the US and EU, are closed.
Regulatory authorities globally demand the incorporation of risk management principles into the life cycle of medical devices.
From the U.S. Food and Drug Administration (FDA) to Australia’s Therapeutic Goods Administration (TGA), and the European Union’s Medical Device Regulation (MDR), a solid risk management system is a prerequisite for market entry.
Risk management is referred to in several places in the MDR and IVDR which apply to the EU market. However, the significantly older 21 CFR 820 for the US market only mentions risk management in the section on design validation. However, the FDA would expect a lot more risk management than the 21 CFR 820 implies because ISO 14971 is an FDA-recognized consensus standard. Furthermore, with the upcoming QMSR, and the inclusion of the ISO 13485 standard in the US regulatory framework, the importance of implementing ISO 14971 principles in the quality management system will increase.
What is ISO 14971?
ISO 14971 is an international standard that establishes requirements for a process for managing risks associated with medical devices. The latest version is the ISO 14971:2019.
There is a European version of the same standard, which is referred to as ISO 14971:2019+A11:2021. The normative parts of this latter standard are the same as in ISO 14971:2019. The forewords and annexes are, however, different.
The A11 annex describes how the requirements of ISO 14971 can be used to demonstrate conformity with the General Safety and Performance Requirements (GSPR) of the MDR and IVDR.
A lot of very important standards in the medical device industry refer to ISO 14971. Thus, it is strongly recommended to implement the requirements of this standard.
ISO 14971 is an example of an external document in ISO 13485, which means you should have access to it if you claim you conform to the requirements. The ISO 14971 can be downloaded from one standard institute that is significantly less expensive than the others.
Standards referring to ISO 14971 include, for example:
- ISO 13485 Quality management systems requirements for regulatory purposes;
- IEC 62366-1 Part 1: Application of usability engineering to medical devices;
- IEC 60601-1 Basic safety and essential performance of medical electrical equipment;
- IEC 62304 Software life cycle processes; and
- ISO 10993-1 Biological evaluation of medical devices within a risk management process.
FMEA and ISO 14971 risk management
FMEA stands for Failure Modes and Effects Analysis. This guide assumes that FMEA has the meaning defined in IEC 60812 and various automotive guidelines on FMEA.
It is essential to note that ISO 14971 product safety risk management is not the same as FMEA. Therefore, these two terms should not be used interchangeably.
FMEA is not required from a regulatory point of view, and the use of FMEA alone does not meet the requirements of ISO 14971, the MDR, or the IVDR.
However, nothing prevents organisations from using FMEA as one tool among others to aid in risk analysis. However, FMEA cannot replace ISO 14971.
Here are some significant differences between ISO 14971 risk management and FMEA:
| ISO 14971 | FMEA |
|---|---|
| Includes both normal and fault conditions, meaning that also side- and after effects are within scope. | Includes only fault conditions. |
| Starts with hazards, which means that risk analysis can start early in any product development process. Hazard identification can be done with very few details about the medical device. | Starts with parts, use, or process steps. This means the design and production processes must be relatively mature before engaging in FMEA. |
| Aims to create a safe medical device. | Aims to create a reliable medical device and production process. |
| Includes both sequences and combinations of events in risk analysis. | Includes only single faults, no sequence or combinations of events. |
| Risk is defined by using the combination of the probability of occurrence of harm and the severity of harm. | Risk prioritisation is often done using the probability of detection. |
| Includes severity of harm when determining the magnitude of risk. | Does not include harm, but only system effects/degradation. |
| Includes not only sequences of events but also combinations of events leading to hazardous situations. | Only includes a single failure event and what it leads to. |
For a more extensive comparison between ISO 14971 risk management and FMEA, see FMEA vs ISO 14971.
Before you start risk management according to ISO 14971
The risk management process
As a manufacturer, you should establish and document a process for risk management. That process should include:
- risk analysis;
- risk evaluation;
- risk control; and
- production and post-production activities.
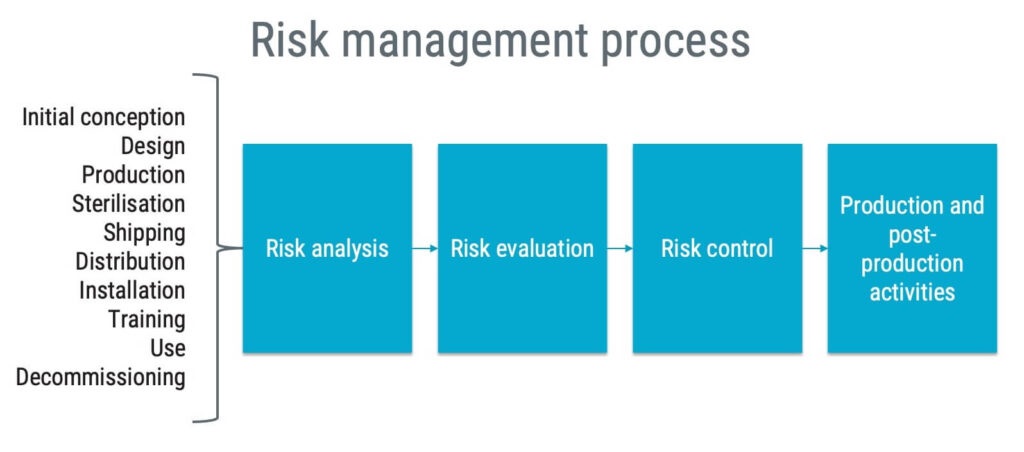
The risk management process should cover the whole life cycle of the device, from initial conception to decommissioning. This means that you should include risks arising from things such as:
- poor design,
- failure in production processes,
- rough handling during shipping,
- software bugs,
- misuse by a user,
- side effects from using the medical device,
- component failures, and
- risks arising during scrapping.
The risks should be taken through the complete risk management process. Please note that your medical device may have different life cycle phases than the example presented above. For instance, not all medical devices are sterile, and sterilisation would, therefore, not be included.
The risk management process must not only be documented, but management should ensure it is effective. Therefore, it is the management’s responsibility to review the suitability of the risk management process at planned intervals to guarantee the effectiveness of it. This requirement should not be confused with risk management review, which is a different activity.
The requirement for risk management training
A basic principle in the medical device industry and quality management in general, is that you should be competent at your job. Requirements relating to this can be found in ISO 13485:
6.2 Human resources
(—)
The organization shall:
a) determine the necessary competence for personnel performing work affecting product quality;
b) provide training or take other actions to achieve or maintain the necessary competence;
The requirement on competence is also present in the ISO 14971 standard:
4.3 Competence of personnel
Persons performing risk management tasks shall be competent on the basis of education, training, skills and experience appropriate to the tasks assigned to them.
… Appropriate records shall be maintained.
Of all the areas that medical device professionals work with, risk management is where auditors are most likely to ask for records of training. It is recommended to have documented training in risk management for at least a few of the persons involved in the process.
In addition to training on risk management, the following knowledge areas must be available when going through the risk management process:
- clinical knowledge;
- application knowledge;
- engineering knowledge; and
- other competencies as needed e.g. microbiologists, environmental engineers, statisticians.
It is a requirement in the ISO 14971 standard to maintain records of who participated in risk analysis and when it was carried out.
Policy for establishing criteria for risk acceptability
One key concept in risk management is whether a risk is acceptable or not. Senior management must provide a policy for establishing criteria for risk acceptability. The policy must be documented and the output from using the policy, i.e. the criteria, should be recorded in your risk management plan. The criteria will be used to determine if a risk is acceptable.
The policy for establishing criteria for risk acceptability should be based on:
- applicable laws;
- relevant international standards;
- generally accepted state of the art; and
- known stakeholder concerns.
For more details and examples, read the article Policy for establishing criteria for risk acceptability.
Intended use and overall design
The intended use is a statement from the manufacturer relating to how the product is intended to be used.
The term intended use is defined in ISO 14971.
Intended use
use for which a product, process or service is intended according to the specifications, instructions and information provided by the manufacturer.
The intended use should answer questions like who would use the device, on whom, for what, where, and when. The intended use is usually found in the instructions for use.
From a risk management point of view, different answers to these questions could significantly impact the risk. For example, is the user a highly trained professional, a layperson, or a child? Will the product be used in a hospital or prehospital setting during emergencies? As such, the intended use must be known when performing risk analysis.
Another precondition for risk analysis is that the medical device must be described in sufficient detail to perform risk analysis. An overview block diagram of the medical device, a written description, or a reference to a similar, previous, or competing product could be used.
As the design matures, the risk analysis should be continuously revisited. New hazards should be added and existing ones refined.
Risk management planning
Risk management planning should occur early in new product development and continue throughout the product’s life cycle. Your plan should be maintained until the shelf-life of your last sold products has expired or the expected service life of your last sold products has been reached.
The risk management plan should at least include:
- the scope of the planned risk management activities;
- assignment of responsibilities and authorities;
- requirements for review of risk management activities;
- criteria for risk acceptability, based on the manufacturer’s policy for determining acceptable risk;
- criteria for accepting risks when the probability of occurrence of harm cannot be estimated;
- verification activities; and
- activities related to the collection and review of production and post-production information.
Risk assessment
Risk analysis is an imperative step for all medical devices. The risk analysis below uses a heart-lung machine as an example. The first step of risk analysis is the identification of hazards, which are potential sources of harm.
The next step involves identifying reasonably foreseeable sequences or combinations of events that could potentially lead to hazardous situations and harm. All this can be documented in a hazard traceability matrix, as shown below.
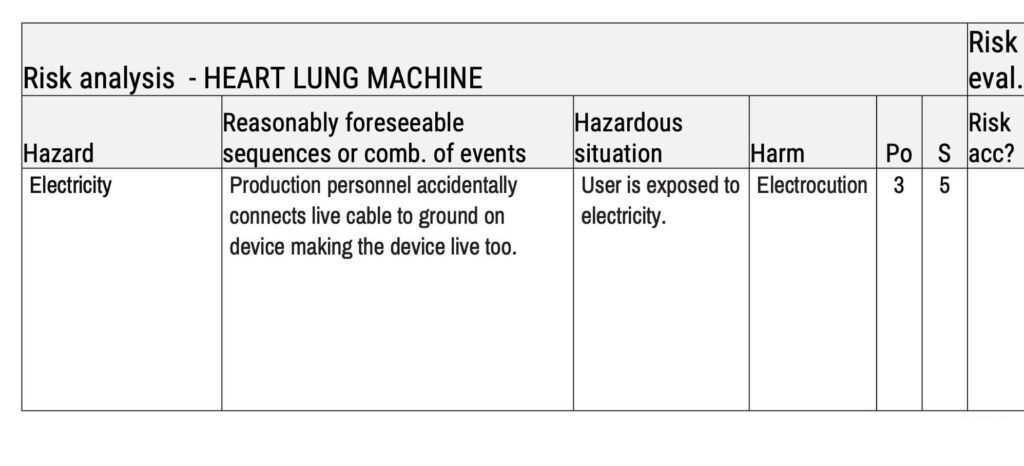
After that, it is time to estimate the risk by determining the probability of occurence of harm (Po) and the severity of that harm (S). In this case, we have used a scale of 1–5 for probability, with 1 being ‘improbable’ and 5 being ‘frequent’. We have also used a scale from 1–5 for severity, with 1 being ‘negligible’ and 5 being ‘catastrophic’.
After you have finished the risk analysis, you perform a risk evaluation, which determines whether the risk is acceptable.
The risk analysis should include risks arising from at least:
- normal use and reasonably foreseeable misuse;
- abnormal use (there are limits to this); and
- technical failures both in the design and the production processes.
What is a hazard?
A hazard is defined as a ”…potential source of harm”. Examples of hazards include:
- electric fields;
- leakage current;
- moving parts;
- suspended mass;
- ionizing radiation;
- bacteria;
- carcinogenic chemical agents;
- particles; and
- irritants.
There are different tools and methods that can be used for identifying hazards, for example:
- answering questions from a checklist with questions relating to the safety of the medical device (ISO /TR 24971:2020 Annex A);
- checking the applicability of hazards from a checklist with hazards (ISO 14971:2019 Annex C);
- brainstorming;
- reviewing previous customer complaints; and
- searching the MAUDE database.
Reasonably foreseeable sequences and combinations of events
After hazard identification, reasonably foreseeable sequences or combinations of events should be identified.
These would depend highly on which type of medical device you work with.
Reasonably foreseeable sequences or combinations of events can come from any life cycle phase and be, for example:
- the wrong fuse is used in production;
- insulation on a cable is broken;
- a software anomaly makes the device behave incorrectly;
- a data breach; or
- a syringe is used on two patients.
How far should you go when you identify reasonably foreseeable sequences or combinations of events?
The standard required you to define the scope of your risk analysis. The scope is generally referred to as what’s included and not, and this could be used to define how far you should go when you identify risks.
Direct and indirect risks from normal use, fault conditions, or reasonably foreseeable misuse should be included. The context in which the medical device will be used should always be considered.
Hazardous situations
After identifying reasonably foreseeable sequences or combinations of events, it’s time to define hazardous situations.
The standard defines the term as:
Hazardous situation
circumstances in which people, property or the environment is exposed to one or more hazards.
This means a few things. First, please note that risk management is not only about patients; it’s about people. This could include users, bystanders, or anyone else. Risk management also includes the environment and property.
Harm
After having identified hazardous situations, it’s time to define harm.
The definition from the standard is:
Harm
injury or damage to the health of people or damage to property or the environment.
Please note that the definition of harm does not limit it to physical harm thus it includes psychological harm.
The important thing to remember here is that it is relevant not only to the patients but also to users and other people. It is a common mistake to exclude risks to anyone other than the patient.
Risk estimation
The last part of risk analysis is risk estimation. It starts with the probability of occurrence of harm (Po).
When estimating Po, you should imagine that you have not implemented any risk control measures for your product; for example, the device has no packaging, casing, insulation on cables, or protection or warnings. It may not be sterilised or even cleaned, and you haven’t considered selecting biocompatible materials.

When estimating risk, be mindful that the probability of harm should be estimated. Do not stop at estimating the probability of occurrence of the hazardous situation but the actual harm.
If Po is based on the probabilities of the events leading up to the hazardous situation. In the flowchart below, the probabilities of Event Pa and Pb are multiplied to get to P1.
P1 is the probability of occurrence of the hazardous situation. P1 is then, in turn, multiplied with P2.
P2 is the probability that the hazardous situation leads to harm. All in all, this will result in Po.
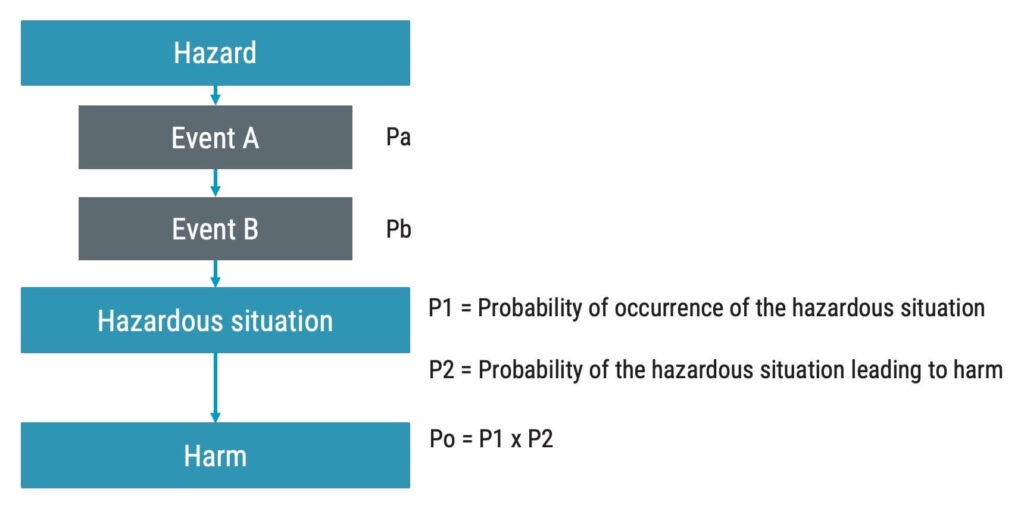
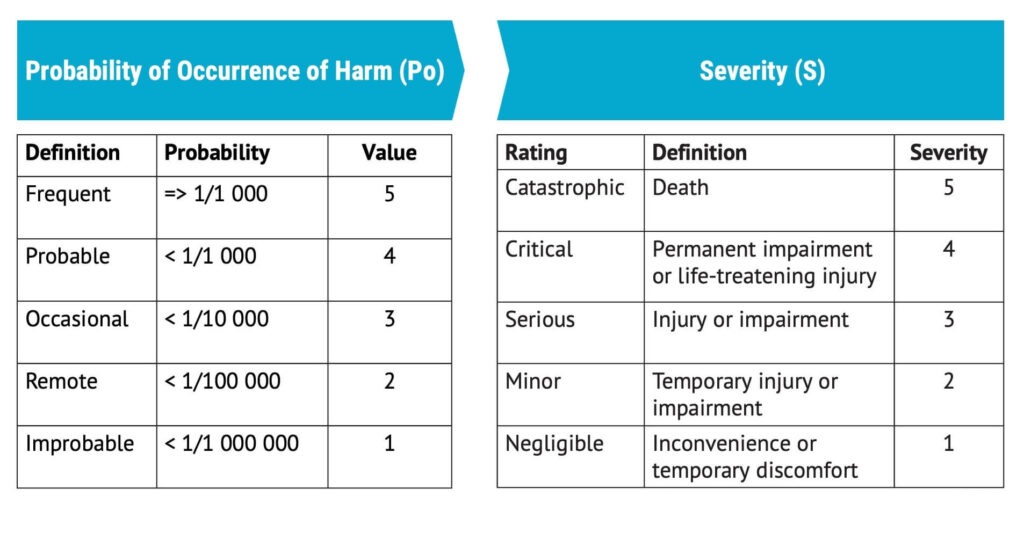
The most common way to assess the probability of occurrence of harm or Po is by measuring it semi-quantitatively so that a certain probability of the harm arising is related to a number. For example, if the probability is greater than 0,01%, that may be represented by a five on a scale from 1–5.
In addition to estimating Po, the severity of harm must be determined.
As in the case of estimating Po, the severity is often estimated using a five-graded scale.
Risk evaluation
One way of performing risk evaluation is to rate the risk on two axes: probability of occurrence of harm and severity of the harm. Below is an example where the green boxes with ACC show an acceptable risk, and the red boxes with N ACC show an unacceptable risk.
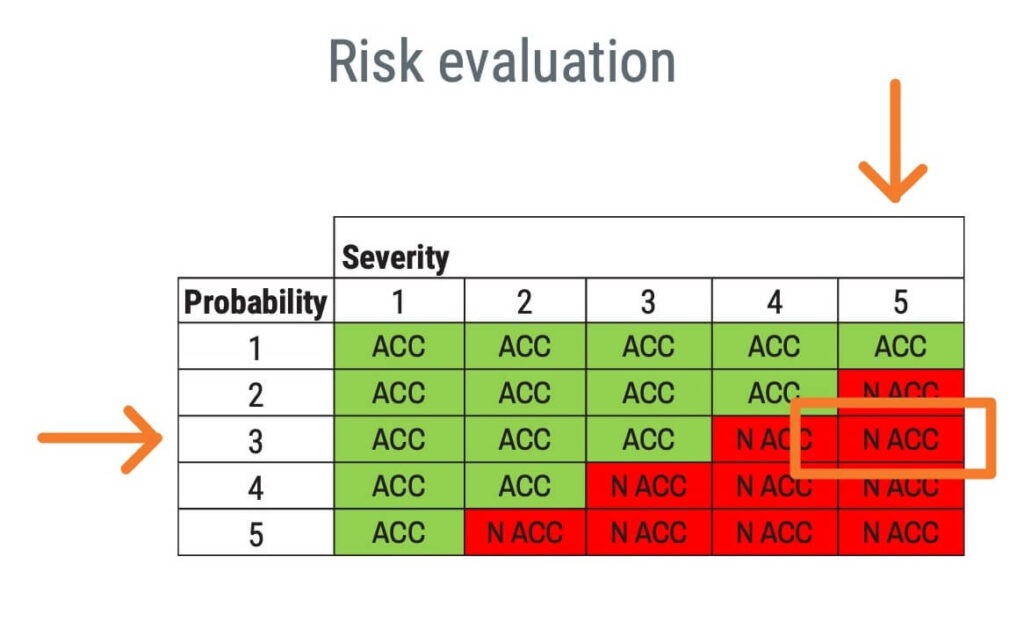
Please note that based on the policy for establishing risk acceptability criteria, other criteria than a risk evaluation matrix may be appropriate.
Risk controls
Following the completion of a risk assessment, should the findings determine the risk to be unacceptable, you must attempt to reduce the risk by implementing risk control measures.
The aim of implementing risk control measures is to reduce risk and ensure that all risks that need to be reduced are effectively reduced. It means decreasing the probability of occurrence of harm or, in some cases, reducing the severity or a combination of the two.
Please note that the severity can also be reduced. It is a common misconception that it cannot be reduced. For evidence of this, see ISO 14971, 7.1:
NOTE 2 Risk control measures can reduce the severity of the harm or reduce the probability of occurrence of the harm, or both.
Risk control options
You can accomplish risk reductions in different ways. There are three different approaches to reducing risk, which are not equally effective.
In the table below, the risk control options from ISO 14971 are shown alongside the risk control options as they are defined in the MDR and IVDR. The risk control options have to be attempted in the order of priority as listed below:
| ISO 14971 | MDR/IVDR |
|---|---|
| 1) Inherently safe design and manufacture. | 1) Eliminate or reduce risks as far as possible through safe design and manufacture. |
| 2) Protective measures in the medical device itself or in the manufacturing process. | 2) Where appropriate, take adequate protection measures, including alarms if necessary, in relation to risks that cannot be eliminated; and |
| 3) Information for safety, and where appropriate, training to users. | 3) Provide information for safety (warnings/precautions/contra-indications) and, where appropriate, training to users. |
Inherent safety by design and manufacture means that you, more or less, remove the hazard or potential source of risk altogether.
Protective measures in the medical device itself or in the manufacturing process aim to prevent exposure to the hazard, although the hazard is still present.
Information for safety or training of users aims to provide users with information or training that will influence their behaviour such that the risk is reduced.
Examples of risk control measures implemented in design and manufacture
| 1) Inherently safe design and manufacture. | 2) Protective measures in the medical device itself or in the manufacturing process. | 3) Information for safety, and where appropriate, training to users. | |
|---|---|---|---|
| Risk control measures implemented in the design | Remove a sharp edge in the design of your product. Remove the welding process used to attach two parts and replace them with one component. Line voltage is replaced with low voltage direct current. | Implement a protective cover that prevents users from pinching body parts. Put bed rails on the patient’s bed to prevent the patient from falling from the bed. Insulation on line voltage cable. | Place warnings on the medical device. Provide instructions for use to support correct use and to avoid use errors. Provide training to users on how to use the device correctly. |
| Risk control measures implemented in the manufacturing process | Remove manual step in production and replace with automation. Remove a toxic release agent from the moulding process. | Perform automated test in production of printed circtuit board assembly to verify production process. Implement an in-process control or final test in production to verify the function and safety. | Not applicable |
Information for safety or user training is a relatively ineffective method of reducing risk. This does not mean it can’t be done, but the effectiveness of the risk control must also be verified when it is information for safety, which is challenging.
Including information safety in the usability engineering process is always recommended. But even if the summative evaluation includes 15 people, it can be challenging to show that the probability of someone not doing something potentially hazardous goes from 0,1% to 0,001% without a significantly larger sample.
Many organisations have historically had this problem. They have reduced a high risk by using warnings but lack the evidence to show that the warning really lowered the risk. For example, they may have asked users if they understood the warning in a questionnaire or checked that the warning is there, but that is not proof of the warning working.
Information for safety vs disclosure of residual risk
There is an important distinction between information for safety and disclosure of residual risk.
“Information for safety” aims to inform the user about what they can do to prevent a hazardous situation or harm from occurring. It is often in the form of a warning, preferably on the device itself.
“Disclosure of residual risk” would refer to the risks you should inform the user about so they can make an informed decision about whether to use the device with regard to the risk of side effects or after effects.

ISO 14971, the MDR, and IVDR will require manufacturers to disclose residual risks.
Risk controls and verification
When working with risk management, you will find that there are at least four main categories of risk controls. Most of your risk control measures will fall within one of these four categories.
- Update the device design to reduce the risk.
- Implement risk controls in your production process.
- Provide training in one form or another.
- Implement a maintenance scheme.
Risk control measures shall be selected and implemented based on the priorities given by the risk control options.
When selecting risk control measures, one should remember that it must be possible to verify the effectiveness of the risk control measures. Several methods can be used for the verification of effectiveness. The method should be chosen based on the risk control measure type and the initial risk.
As a general principle, if the magnitude of risk is low before risk control measures, less time and rigour are needed to verify the effectiveness.
The corollary is also true; the more significant the magnitude of risk is in the risk analysis, the more time and rigour should be applied to verify the effectiveness.
Exceptions to the principles above are if the design or manufacturing process has been made inherently safe, then it may be sufficient just to conclude that the hazard has been removed.
ISO 14971 requires both the effectiveness and implementation of the risk control measures to be verified, and records must be maintained of both.
What is the difference between verification of effectiveness and verification of implementation?
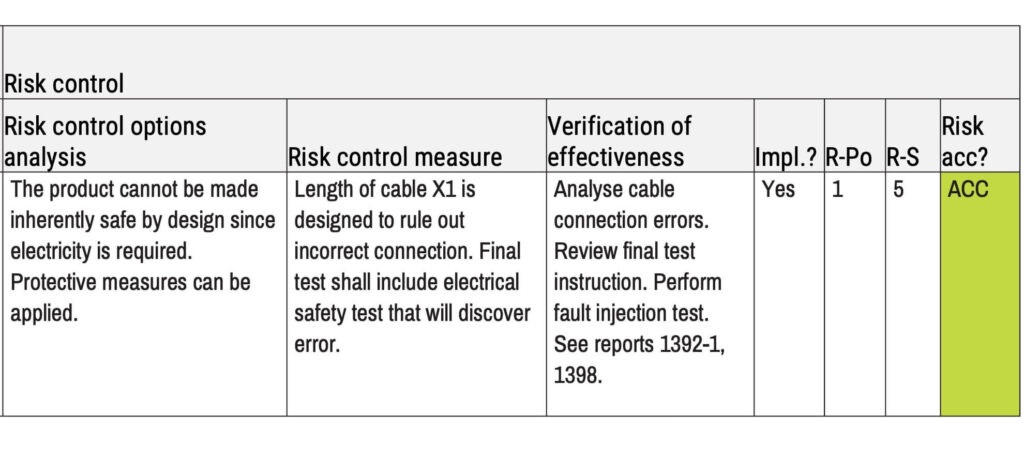
Verification of effectiveness and implementation of risk controls are often mixed up. Here is an example of how they are different and what can go wrong:
During the usability engineering process, the usability engineers identify a risk where the risk control measure should be a warning on the medical device itself.
The engineers created and attached a warning label to the products used in the summative evaluation. The evaluation demonstrates that the warning label works. However, as the engineers added the label at a late stage, it was not added to the manufacturing specifications. Thus, the warning has been proven to work (verification of effectiveness), but it was not implemented.
The risk management standard ISO 14971 and its technical report ISO/TR 24971 mention reviewing production specifications to verify the implementation.
I recommend taking the verification of effectiveness one step further by practically inspecting the production line to ensure that the risk controls that should be implemented in the design have been put into production and that the risk controls that should be implemented in the production have been implemented.
In this example, the verification of implementation is found in the column marked “Impl.?”.
Using standards as risk controls
If a standard applies to your medical device, it should meet at least the requirements or safety principles in the standard.
There are several links between standards and safety. Safety is freedom from unacceptable risk. Standards are supposed to represent state of the art, which is defined in the risk management standard as:
State of the art
developed stage of technical capability at a given time as regards products, processes and services, based on the relevant consolidated findings of science, technology and experience
Note 1 to entry: The state of the art embodies what is currently and generally accepted as good practice in technology and medicine. The state of the art does not necessarily imply the most technologically advanced solution. The state of the art described here is sometimes referred to as the “generally acknowledged state of the art”.
When reviewing the two definitions, you can easily draw a line between “acceptable risk” and “generally accepted as good practice in technology and medicine.” If the requirements of the standard are met, the magnitude of risk is likely to be at a level that is generally accepted as good practice in technology and medicine, which should be equivalent to acceptable risk. This is not always true, but it can be used as a rule of thumb.
Standards that help with risk management fall into two main categories:
- The standard provides design inputs, risk controls, and the verification method to show effectiveness.
- The standard provides the only verification method.
It is essential for any medical device development effort to identify applicable standards as part of the regulatory strategy. The standards shall also serve as input to risk analysis, the selection of risk controls, and the verification of their effectiveness.
What are the risks arising from risk control measures?
Please note that sometimes, when you introduce a risk control measure, you also introduce new risks. Always review risk control measures to ensure new hazards are not introduced or existing ones are changed.
If a new risk is introduced, the risk is to be treated just like any other risk, meaning that you add it to your hazard traceability matrix.
Estimating the residual risk and benefit
Residual risk estimation
Estimating the residual risk is done the same way as estimating the risk in risk analysis. However, the residual risk relates to the risk that remains after implementing risk control measures. As mentioned above, please note that risk control measures can reduce Po, severity, or both.
The residual Po and S are often referred to as R-Po and R-S.
For risks that don’t meet the risk acceptability criteria or where there are no available risk control measures, you must perform a benefit-risk analysis.
There are more considerations when planning on placing the medical device on the EU market.
Residual risk in the EU
When looking at the MDR and the European Union, it’s a bit more complicated than previously mentioned.
Firstly, the MDR and IVDR require performing benefit-risk analysis for all the individual risks. This means not only the risks where residual risks are unacceptable but all risks.
From one point of view, there is nothing strange about this. Everyone working on the design of a medical device or production process weighs the benefits against the risks all the time.
The question is how much of the benefit-risk analyses should be documented. Regarding this, different auditors will have different views. To avoid nonconformities, reference some kind of benefit-risk analysis for all individual risks.
Evaluation of the overall residual risk
There are two primary purposes for the overall residual risk evaluation.
- Ask yourself on a high level whether the product’s overall residual risk should be considered acceptable. This is really a question of whether your product is better or worse or on par with every other product in the field.
- Ensure that you compile and communicate the residual risks of your medical device.
One way to understand overall residual risk acceptability is to compare two products with two graphical representations of their risk profiles:
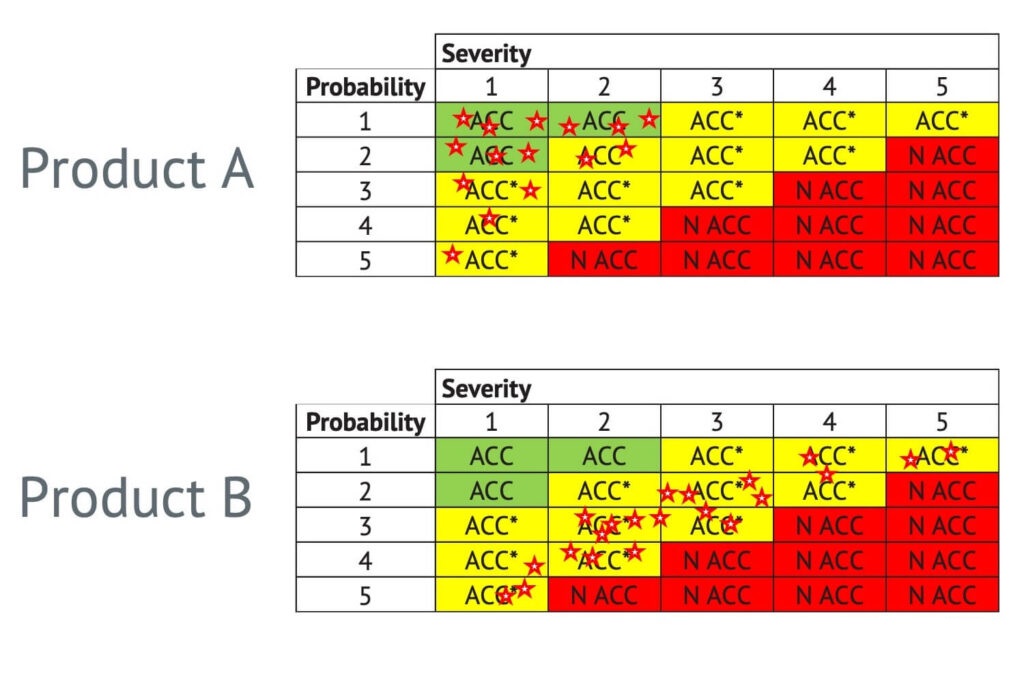
Product A has a more favourable risk profile compared to product B.
How to evaluate the overall residual risk
You could compare the risks associated with your product with competitors, have application specialists or experts review your product, or use expert judgment. All three methods overlap in one way or another.
I recommend that you perform a qualitative analysis similar to the kind of discussion required to be documented in your clinical evaluation report, according to MEDDEV 2.7/1 rev 4.
Risk management review
The risk management review should address at least three things.
- Was the risk management work executed according to the plan, that is, the risk management plan and any applicable SOPs?
- Is the overall residual risk acceptable?
- Are appropriate methods in place for collecting production and post-production information?
Writing the risk management report is one of the last things within risk management you will complete before placing the medical device on the market. This will often take place in conjunction with design validation. And you may wonder why it would be so late in the project. That is because some of the last risks you’ll find will likely be identified during design validation when you see real people using your medical device.
Please note that top management must review the suitability of the risk management process at planned intervals. This review is not the same as a risk management review.
A risk management review is typically done at the end of a product development project, whereas the suitability of the risk management process is a continuous activity.
The risk management file
The last general requirement mentioned in the standard is that the manufacturer establish and maintain a risk management file. It compiles all the documents created during the risk management process. It may be a binder, physical or electronic, containing all the documents or a list referencing the results of risk analysis, evaluation and control, and the risk management report.
This file should be maintained as long as the product is in use and should provide the information necessary to review the risk management process at any phase in the medical device’s life cycle.
The risk management file should at least include:
- preliminary hazard analysis (or equivalent;
- risk management plan;
- characteristics related to safety incl. limits;
- hazard identification list;
- hazard traceability matrix;
- risk management report;
- software safety classification (when applicable); and
- risk management review (design review).
Production and post-production activities
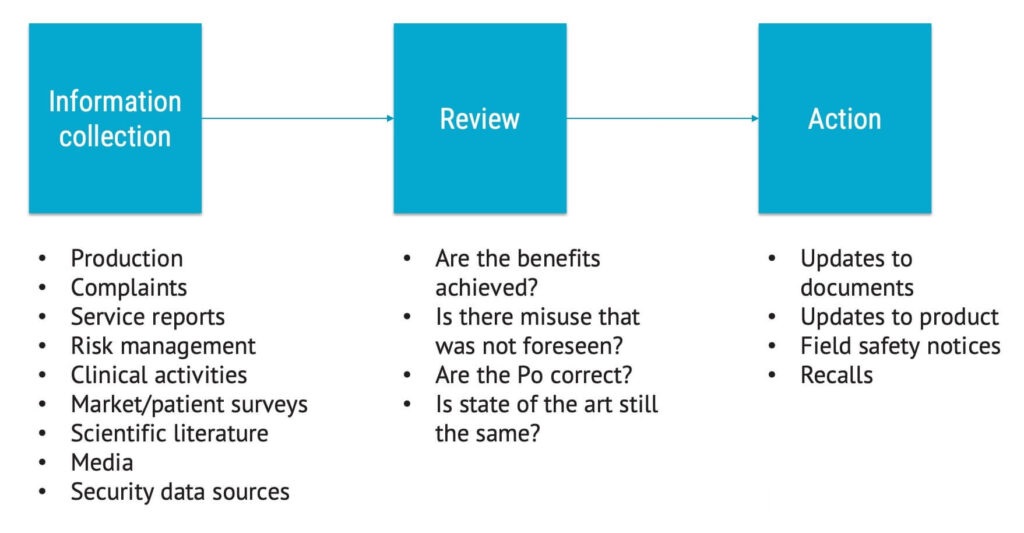
In the image below, you can see how production and post-production activities feed back information into the risk analysis, risk evaluation, and risk control process, which should happen repeatedly over time.
On a side note, the section on production and post-production activities has been significantly expanded in the 2019 version of the standard compared to the last version, which was not as comprehensive in this field. It’s an excellent change and update.
The process of production and post-production activities is shown below with example questions.
Making this process work is not simple. If you’re a small manufacturer, you might struggle with having the administrative systems in place to ensure that you collect all the information. If you’re a large company, you might struggle to get every customer service representative to report what they learn about risk in the right way and the right place.
Summary: Working with risk management for medical devices and ISO 14971
Working with risk management and having a process or a system for it is required for all major medical device markets worldwide. ISO 14971 is the go-to standard for this purpose.
Risk management comprises planning, risk analysis, risk evaluation, risk control, overall residual risk evaluation, risk management review, and production and post-production activities.
FMEA does not, on its own, meet the requirements for risk management and ISO 14971.
It is a requirement that participants performing the risk management work are trained in risk management and have certificates to prove it.